| WHY? | HOW? | WHAT? | WHAT NEXT? |
The details of the mechanism of the dissociative chemisorption of methane
on transition metal surfaces have been extensively studied both experimentally
and theoretically due to the relevance of this process to catalysis, and
in particular, as a model for C-H bond activation on metal surfaces. Methane
dissociation is a direct, activated process on both platinum and nickel
surfaces in which the sticking probability increases with increasing incident
translational energy.
An original qualitative model for the reactivity that was proposed involved thermally assisted tunneling through the reaction barrier which was based on the positive dependence of the sticking coefficient with increasing surface temperature and the measured kinetic isotope effect. The chemisorption mechanism is, however, not yet fully understood with respect to the dependence of the sticking coefficient on the different forms of energy involved in the reaction, including translational energy, thermal surface energy, and the internal energy of the methane molecule. For instance, since it is known that the barrier for reaction occurs on the exit channel, vibrational excitation of the incident methane should significantly increase the sticking probabilities.
What is not yet known is the relative efficacy of stretching or bending vibrational modes in the promotion of the dissociative chemisorption. To gain additional insight into the reaction mechanism and the state selective dependence on vibrational energy, we perform experiments that measure the dissociative chemisorption of methane on transition metal surfaces (Pt and Ni) prepared in single ro-vibrational states with well defined translational energy.
To measure the quantity of methane that sticks to a metal surface and
hence determine the sticking coefficient at zero coverage, we employ the
technique of thermal energy atomic scattering (TEAS). The specular reflection
of a thermal energy helium beam from a clean metal surface is very high
and can be monitored using a mass spectrometer. As the methane
molecules stick to the surface, the specular intensity decays because some
of the He atoms are scattered off the specular direction by the surface
irregularities generated by the chemisorbed molecules. Since helium/methane
mixtures are used in the experiment, the coverage of methane on the platinum
surface can be monitored continuously throughout the experiment by tuning
the mass spectrometer to m/z=4 as the crystal surface is exposed to the
helium/methane from the molecular beam. TEAS is very sensitive to small
coverages due to the large cross section for surface scattering of helium
from absorbed molecules. In this way, we are assured that the sticking coefficients
that are measured are a true representation of S0, since only
the earliest fraction of the He specular decay which corresponds to very
low coverages are used in the determination of S0.
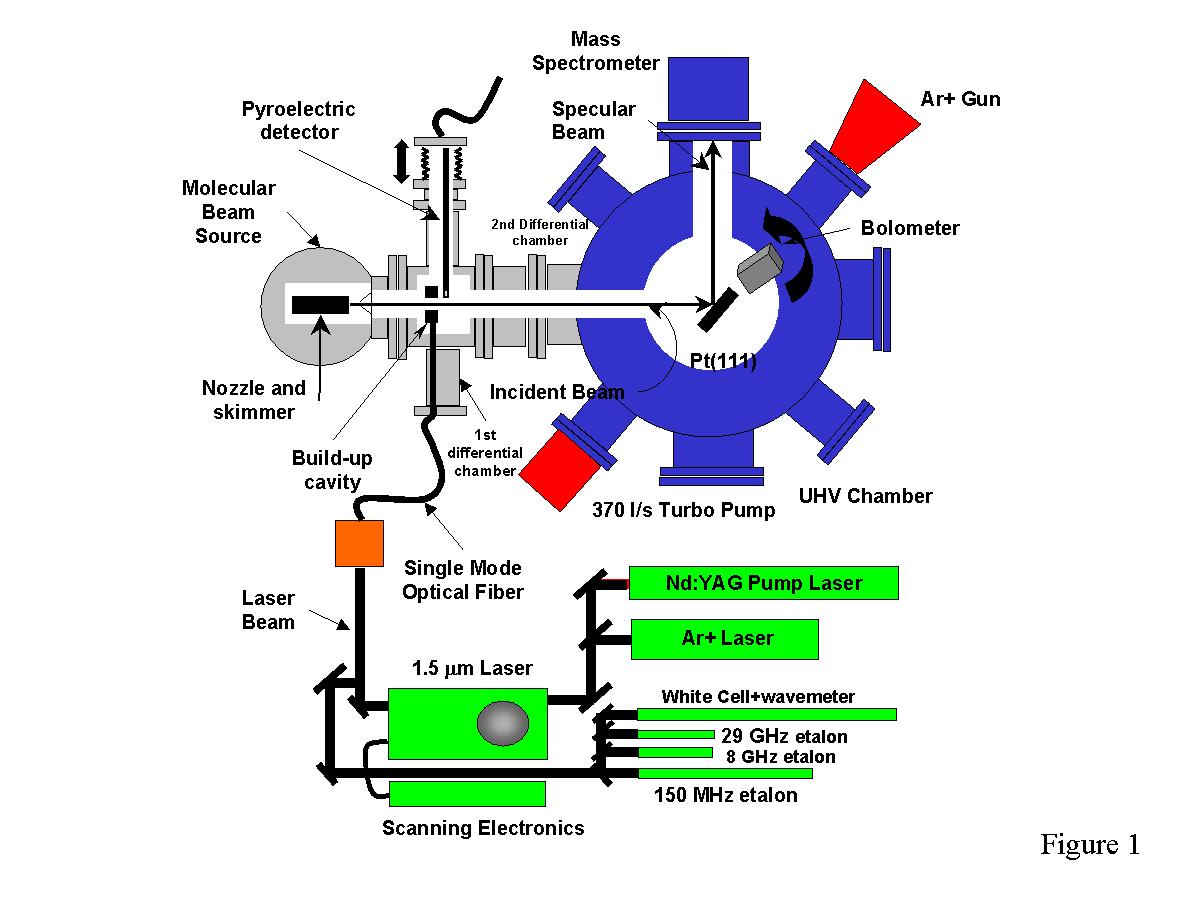
The experimental apparatus consists of a UHV chamber coupled with a molecular
beam source (a schematic of the experimental apparatus is shown in Figure
1). The UHV chamber where the platinum crystal is located is pumped by a
370 l/s turbomolecular pump and has a typical base pressure of 3x10-11
torr. The molecular beam source is pumped by a 3000 l/s
diffusion pump and consists of a supersonic expansion that is produced by
expanding a high pressure gas mixture through a 23 micron quartz nozzle.
The nozzle can be heated from 300K to 1100K, which along with changing the
ratio of methane to helium, allows the adjustment of the translational energy
of the methane in the beam. The chopped molecular beam is skimmed and passes
through two turbomolecular-pumped chambers before entering the UHV chamber
where it impinges on the crystal that is mounted on a six axis manipulator
in the center of the chamber. A quadrupole mass spectrometer with an axial
ionizer is mounted perpendicular to the molecular beam direction and monitors
the He specular reflection at thetai=thetaf=45 degrees.
After cleaning the crystal surface, the specular intensity of pure He is
monitored for several hours to insure that the surface remains clean.
The methane molecules in the beam are vibrationally excited through the
use of a continuous wave (CW) 1.5 micron Burleigh color center laser that
is operated on a single longitudinal mode. The laser is actively stabilized
to a 150 MHz external etalon using an intracavity electro-optic crystal.
The linewidth of the laser is determined to be on the order of 1 MHz. The
laser
radiation is transported to the molecular beam apparatus by way of a single
mode optical fiber. The experiments consist of measuring the slope of the
specular decay produced by the sticking of the excited methane and comparing
it to the slope obtained when unexcited methane is adsorbed.
The translational energy dependence of the sticking coefficient is depicted in Figure 2 with a comparison to the data of Luntz and Bethune [1]. The platinum surface was maintained at 575 K in our experiments while those of Luntz and Bethune were obtained at Tsurf=800 K. If our measured surface temperature dependence of the sticking coefficient is taken into account, our data shows good quantitative agreement with that of Luntz and Bethune in the translational energy range of 20-55 kJ/mol.
Having established that the translational energy dependence of the sticking coefficient can be reliably measured using TEAS, we performed experiments where the laser was both on and off resonance with a transition to the 2n3 level while the molecular beam of CH4/He was impinging on the crystal. A beam of 80% CH4/20% He was used for the experiments in which laser excitation of the methane was performed. The nozzle was kept at 295 K which gave a normal translational energy of 5.4 kJ/mol. Figure 2 also displays the sticking coefficient of the molecules without laser excitation (blue triangle) providing the control experiment to verify the proper functioning of the experimental apparatus. Experiments were performed by tuning the laser to both the Q(1) line (6004.827 cm-1) and the R(1) line (6026.208 cm-1). By using these two lines, we could pump both the J=1 and J=2 rotational states of the excited vibrational level. In the analysis of the data, the effect of the rotational state will be neglected since the energy difference is small in comparison to the vibrational energy and hence a change in sticking coefficient was not detected within the errors of our experiment.
The mean sticking coefficient S0 of the control experiments
without laser excitation was determined to be 6x10-6 at 5.4 kJ/mol
normal translational energy. The total sticking coefficient derived from
the specular decay curves of the experiments where the laser was exciting
the transition was determined to be 2x10-5 included the contribution
from both laser excited and unexcited methane. The state selective sticking
coefficient of methane in the 2n3
J=1,2 states could be found by taking the difference between the control
experiments and those with laser excitation and normalizing to the fraction
of molecules excited in a given experiment. By following this procedure,
S0 of the 2n3 J=1 and J=2
state of methane on Pt(111) is found to be 1.8x10-4 (Figure 2
red square). This represents an enhancement factor in the reactivity of
28-fold for methane with two quanta in the asymmetric stretch as compared
to the reactivity of the ground vibrational state. Even at internal energies
higher
than the barrier for dissociation, the efficacy of the vibrational energy
is only 40%. This points to the fact that a truly statistical picture of
the reaction mechanism is not adequate, as the total energy in the collision
should be enough to overcome the barrier with near unit probability.
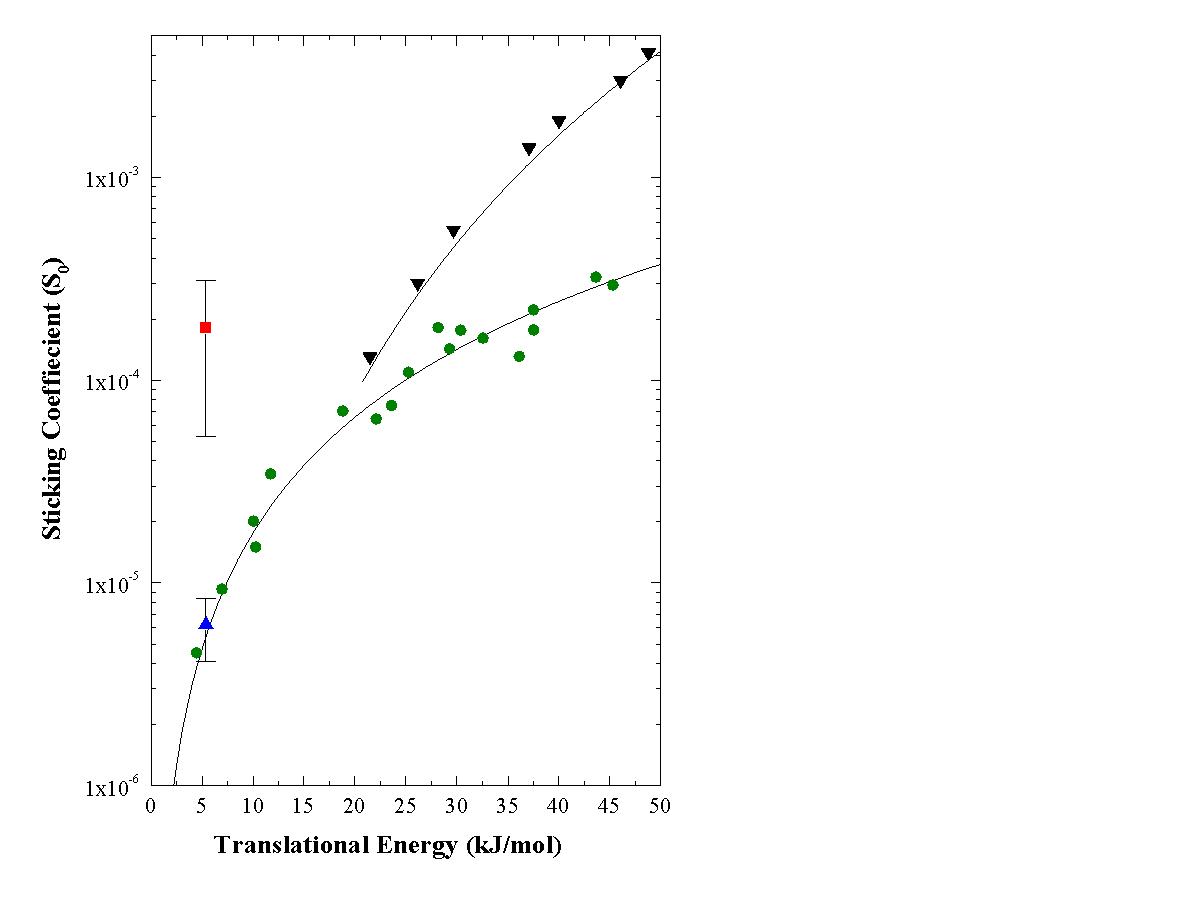
Figure 2. Comparison of the sticking coefficient of the 2n3 state of CH4 (red square) with the translational energy dependence obtained without laser excitation (green circle) on Pt(111) . The sticking coefficient of the control experiment at 5.4 kJ/mol is given by (blue up triangle). The error bars are 2sigma (95% confidence limits) of replicate measurements. Measurements obtained in this work using TEAS at a surface temperature of 575 K, (black down triangle)-data obtained from Luntz and Bethune at a surface temperature of 800 K [2].
Experiments are currently being carried out in our laboratory to measure the vibrational activation of methane chemisorption on the (111), (100), and (110) faces of nickel. In the only other experiment of this kind ever reported, Utz and co-workers [2] have shown that the enhancement factor for excitation of the 1n3 vibrational level is greatest at the lowest incident translational energies. Our experiments on Ni should provide information on the dependence of the reactivity of methane on nickel as a function of the extent of the vibrational excitation.
We are also interested in pumping different modes of methane and determining
how the sticking coefficient changes. In this way, we can determine if bending
or stretching modes (or combinations thereof) are more effective at promoting
chemisorption at energies comparable to the barrier height for dissociation.
These studies can also be extended to include
other small molecules that have a low sticking coefficients on metal surfaces.
Saturated hydrocarbons, both alkanes and those with functional groups, are
good candidates for these studies while unsaturated hydrocarbons have been
found to dissociatively chemisorb on Pt and Ni with high probabilities,
irrespective of their vibrational excitation.
Finally, we are presently studying, using again He atom reflectivity as a detection method, the resurfacing of H atoms which have been buried under the surface by bombardment with 5eV Xe atoms. Subsurface hydrogen is an important and not well known factor in many surface chemical processes.
[1] A. C. Luntz and D. S. Bethune. J. Chem. Phys. 90, 1274
(1989).
[2] L. B. F. Juurlink, P. R. McCabe, R. R. Smith, C. L. DiCologero, and
A. L. Utz. Phys. Rev. Lett. 83, 868 (1999).