5. Activated Chemisorption of Methane
Since the invention of the laser, researchers have been fascinated with the possibility of promoting chemical reactivity by the excitation of vibrational modes in molecules. By using finely-tuned photons instead of traditional broad spectrum heat sources, the rates of kinetically-limited reactions could be potentially enhanced while the rates of competitive or impurity generating processes remained low. Although early experiments were often attempts to induce reactivity by vibrationally exciting a diatomic molecule closer to its dissociation limit, this field has expanded considerably with advances in laser technology and methodology. At present, researchers are exploring the role of vibrational energy in the determination of branching ratios between reactive pathways, rates of isotope-selective unimolecular cleavage, and the promotion of adsorption onto reactive surfaces.
In this chapter, the effect of vibrational energy on the rate of adsorption of methane on Pt(111) and Ni(111) surfaces is explored. Using the external resonant cavity technique of molecular beam excitation developed by Scoles and coworkers, methane in a molecular beam was excited with two quanta of the 3 asymmetric stretch before encountering the surface. By performing direct laser excitation of a significant fraction of methane in the beam, mode-specific measurements of any potential change in rate of adsorption were possible.
5.1.1. Gas-Phase Reactivity
The effect of internal energy on chemical reactivity was extensively explored by the work of Polanyi. To model the gas-phase exchange reaction between a neutral atom (A) and a diatomic molecule (BC) to produce a new species (AB), potential energy surface diagrams were developed which describe the energetics of the reaction as a function of the interatomic distances. For many reactions, the presence of an activation barrier will prevent the reaction from proceeding without sufficient free energy. However, the location of this barrier in either the entrance channel (an "early" barrier, Figure 5-1a) or the exit channel (a "late" barrier, Figure 5-1b) will determine the efficacy of vibrational energy for promotion of the reaction.
Figure 5-1. Contour plots
of potential energy surfaces from Reference 7: a) Early barrier,
b) Late barrier. Vibrational energy is particularly effective
for late barrier reactions since the bond length in the original
molecule must be increased for dissociation to occur with a minimum
of energy.
The effect of vibrational energy can be explored by plotting reaction trajectories using the two types of potential energy surfaces shown in Figure 5-1. If the activation barrier is located in the entrance channel of the reaction, a translationally excited molecule will be able to encounter and optimally travel across the saddle point of the activation barrier. If the molecule is vibrationally excited, the probability of encountering the local minimum of an early barrier is reduced; however, the presence of additional energy may still increase the probability of a successful crossing at a location that is less than optimal. Alternatively, if the activation barrier is located in the exit channel, the bond-length oscillation induced by vibrational excitation will enable the intramolecular distance to increase so that the molecule can cross the barrier at the saddle point. In this "late" barrier case, vibrational energy not only provides additional energy to overcome the activation barrier, but also couples to the reaction coordinate much more efficiently than translational energy.
The enhancement of gas-phase exchange reaction rates
by vibrational excitation of the reactants was measured by crossed
molecular beam experiments which used lasers to excite the stretching
vibration in a HCl molecule prior to a reactive collision with
a neutral atom. One quantum of vibrational energy (h=34.5 kJ/mol)
increased the rate of reaction with potassium and oxygen atoms
by a factor of 100 and 400, respectively. In addition, two quanta
of vibrational excitation (h=67 kJ/mol) prior to the reaction
of HCl with a bromine atom increased the rate of reaction by a
factor of 1011.
5.1.2. Internal Energy and Adsorption
To address the effect of internal energy on the rate of adsorption at surfaces, independent sets of theories are available for the two basic processes of adsorption: physisorption and direct activated chemisorption.
For physisorption, the probability of adsorption is determined by the translational energy of the impinging molecule after the surface encounter. If the molecule loses enough translational energy during a collision with the repulsive wall of the surface, the molecule will be unable to escape the potential well. Any conversion of internal energy to translational energy during the surface encounter will potentially reduce the probability of physisorption. Early predictions of this effect by Kochelashvili, et al. were based on an equal contribution of all energy modes, both internal and translational. In this simple model, all energy in the molecule was mixed upon collision with the surface. The probability of sticking was then determined by an Arrhenius relationship as a function of the total energy of the molecule and a critical energy parameter. Later studies by Karlov and Shaitan refined the model by quantitatively accounting for the vibrational to translational energy transfer for light and heavy incident molecules. Further improvements by Doll predicted that only internal modes which were low in energy (e.g. rotations and slow vibrations) would decrease the probability of adsorption. Recently, molecular beam studies of the physisorption of SF6 and CCl4 have shown that the rate of internal to translational energy conversion is relatively small but may be rate-determining in the limit of low translational energy.
In contrast, for activated chemisorption processes, energy is required to overcome an activation barrier to adsorption. As a result, the presence of energy in internal modes will potentially increase the likelihood of chemisorption. Simulations by Gelb and Cardillo of the dissociative chemisorption of H2 on Cu(111) determined that the probability of dissociation increased by a factor of 42.5 with one quantum of vibrational energy. Similarly, one quantum of vibrational excitation in D2 resulted in a 63-fold increase in reactivity. With more sophisticated models to account for non-ideal experimental conditions (e.g. experimental spread of the incident velocities), Darling and Holloway have reexamined the rate of chemisorption of H2 on a variety of transition metal surfaces. The comparison of these molecular simulations to experimental results has confirmed that the location of the activation barriers, early vs. late, will strongly determine the dependence of the adsorption rate on vibrational energy. However, in spite of recent advances in model design and computing power, most simulations continue to be limited to the examination of vibrational effects in diatomic and quasi-diatomic molecules.
5.1.3. Early Adsorption Experiments
Studies of the dissociative chemisorption of methane on tungsten provided the first indirect observations of an adsorption rate dependence on the degree of internal excitation. Using heated tungsten filaments maintained under low vacuum, Winters observed that the rate of chemisorption of CD4 was an order of magnitude less than that of CH4. Since this difference in rates decreased as the surface temperature increased, a tunneling mechanism along the C-H vibrational stretch coordinate was proposed. In this model, the vibrational excitation of methane was generated by the thermal equilibration of the molecule with the hot surface while residing in a short-lived precursor state. At lower surface temperatures, only the lighter hydrogen atoms could tunnel through the activation barrier to dissociation while the deuterium atoms could not. A similar isotopic dependence of the rate of adsorption of deuterated methane species on rhodium was observed by Stewart and Ehrlich. By systematic elimination, only the vibrational excitation of methane was determined to be responsible for the observed rate differences. Electronic excitation was too high in energy to readily occur, rotational modes were too low in energy to significantly contribute, and translational energy could not account for the observed isotope effect.
To directly measure the effect of vibrational energy on the rate of methane chemisorption, the research groups of Ehrlich and Yates and Weinberg each used a laser to excite the 3 and 24 modes of methane molecules in proximity to a rhodium surface. While the crystal was exposed to a residual pressure of two Torr of methane, a He-Ne laser provided up to 38 mW of photons to excite the P7 transition of the 3 asymmetric stretch of methane at 2947.906 cm-1. Methane molecules excited in the 24 mode were generated by deexcitation of the 3 mode during collisions with helium atoms. If successful, the dissociative chemisorption of methane would have been detected by an increase in the partial pressure of hydrogen gas in the reaction chamber. However, low laser power combined with large fluctuations of the hydrogen partial pressure in the chamber prevented the detection of any change in the rate of methane chemisorption due to laser excitation.
Although adsorption experiments performed with vibrationally excited methane were inconclusive, researchers have successfully promoted other surface reactions with the use of laser radiation. The rate of dissociative chemisorption of nitrous oxide on copper was enhanced by a factor of 5000 with the excitation of the 3 asymmetric stretching mode. Similarly, the rate of ammonia decomposition on platinum was increased by several orders of magnitude by the addition of one quantum of vibrational energy. However, for both studies, control experiments with the laser frequency detuned off of the transition were not performed. As a result, it is not known if the reported increases in adsorption rate are due to mode-specific or thermal effects.
Branching ratios were also shifted by vibrational
excitation of the reactants for adsorbate/substrate systems with
multiple surface reaction pathways. In the unimolecular decomposition
of formic acid on platinum, the CO2:CO product ratio
was doubled during laser excitation. Irradiation of NO2
molecules before reaction with ethylene in the presence of a platinum
catalytic surface increased the CO2:CO product ratio
by 50%. In each study, the product ratio depended on the absorption
coefficient of the transition chosen for laser excitation. For
weakly absorbing bands, the product ratio enhancement was less
than that of more strongly absorbing bands. However, due to the
high probability of collision induced vibrational deactivation
or conversion, the specific vibrational mode responsible for the
observed behavior could not be determined.
5.1.4. Molecular Beam Experiments
Although laser-based experiments provided evidence that vibrational excitation could be effective for promoting reactivity at surfaces, these experiments were limited to molecules with strong absorption features. For other systems, a population of vibrationally excited molecules is generated by a supersonic expansion from a heated nozzle. With few molecular collisions in the expansion, the vibrational temperature of the molecular beam remains approximately equal to the nozzle temperature. With the vibrational modes populated according to Boltzmann statistics, several experiments performed at different nozzle temperatures could potentially determine the relative contribution of each mode to the observed rate of adsorption.
Using this technique, several activated adsorption systems were studied by Rettner and coworkers. Initial studies of the chemisorption of methane on tungsten and rhodium surfaces determined that vibrational energy was potentially as effective as translational energy for promoting reactivity. However, due to the large number of vibrational modes of methane which were populated at high nozzle temperatures, the efficacy of any particular mode could not be determined. From the study of H2 and D2 adsorption on Cu(111) (molecules with only one vibrational mode available), the vibrational energy of the stretching mode was as effective as translational energy for the promotion of chemisorption,. In contrast, vibrational energy was only half as effective as translational energy in studies of the dissociative chemisorption of N2 on Fe(111). For both systems, the incremental enhancement of the rate of adsorption per quantum of vibrational energy diminishes with increasing excitation.
To simplify the computations required to analyze
the adsorption of molecules with multiple vibrational modes, quasi-diatomic
potentials have been developed for molecules such as methane.
Instead of considering all possible modes of the original molecule,
a hypothetical CH3-H molecule is created with a single
stretching mode with a vibrational energy of approximately 3000
cm-1 per quantum. Using this technique, no mode-specific
information is available; however, visualization of the data becomes
comparable to that for a true diatomic like H2 or D2.
With this model, vibrational energy was observed to enhance the
rate of chemisorption of methane on nickel slightly more effectively
than a comparable amount of translational energy29.
5.2. Methane Adsorption on Transition Metal Surfaces
5.2.1. Nickel Surfaces
The dissociative adsorption of methane on nickel catalytic surfaces is the rate-limiting step for the steam reforming process, CH4 + H2O CO + 3 H2. This process is used to generate compounds which are used as feedstocks for the production of synthetic fuels, polymer resins and related chemicals. The activation energy for direct dissociative adsorption of methane on a Ni(111) single-crystal surface is 52.7 kJ/mol.
Measurements of the rate of adsorption of methane on low-index nickel planes previously had been significantly confused by the existence of a "pressure gap". Reports of sticking coefficients measured under conditions encountered in industrial reactors or at high pressures (10-2-1000 Torr) differed significantly from sticking coefficients determined under high vacuum conditions ( 105 Torr). Initial explanations of the anomalous adsorption rates were based on the deformation and vibrational excitation of methane during collision with the surface at high translational energy. The lower-energy bending modes of methane (2, 4) would be expected to be populated most by the conversion of translational energy to vibrational energy. The resulting deformation of the molecule during the surface encounter would expose the central carbon atom to the surface, increasing the probability of dissociative chemisorption.
This collision-induced deformation could be simulated by exciting vibrational modes in impinging molecules. Although the bending modes provide significantly greater exposure of the carbon atom to the surface, stretching modes could also disturb the molecular geometry sufficiently to increase the likelihood of chemisorption.
5.2.2. Platinum Surfaces
The adsorption of methane on platinum surfaces is qualitatively similar to adsorption on nickel surfaces. However, the activation barrier to dissociative chemisorption is approximately 10-20 kJ/mol less than that observed on equivalent nickel surfaces.
In a study of adsorption on Pt(111), Luntz and Bethune observed that the dissociative chemisorption barrier could be overcome by increasing the translational or internal energy of the incident methane, or by raising the temperature of the platinum crystal. However, the relationship between the sticking coefficient and the translational energy of the incident molecule along the surface normal was not strictly exponential, potentially indicating the presence of multiple barriers or alternative pathways to adsorption. In addition, there was a strong dependence of the sticking coefficient on surface temperature which could be explained with an adsorption mechanism of "thermally assisted tunneling". During collision with the surface, the molecule could utilize thermal energy from the surface to promote dissociation at a rate faster than predicted by direct adsorption models.
If the tunneling model presented in the literature
is valid, the excitation of stretching vibrational modes would
increase the likelihood that the carbon-hydrogen bond length would
be large enough for tunneling to occur.
In view of the literature reports of the enhancement
of the rate of adsorption by vibrational excitation of the impinging
dose molecules, we became interested in performing direct laser-excitation
experiments to test the mode-specificity of this effect. With
the high intracavity power density generated by an external resonant
cavity, an appreciable fraction of methane could be, for the first
time, excited. In addition, with the high sensitivity of helium
atom reflectivity (conveniently performed with the same molecular
beam used to excite and deliver the methane to the surface), very
small rates of adsorption could be measured with the goal of observing
relative changes in the sticking coefficient due to vibrational
excitation of the incident methane molecules.
5.3. Results
The rate of methane adsorption was determined from helium atom reflectivity measurements during exposures of a Pt(111) or Ni(111) crystal to various molecular beam concentrations of methane seeded in helium (0.510%). The translational energy of the incident methane molecules was varied from 12 to 40 kJ/mol by adjustment of the nozzle temperature. To minimize the rate of contaminant adsorption, the surface temperature was maintained at 600 K during deposition for most experiments. Sticking coefficients were determined as the ratio between the rate of methane adsorption divided by the methane flux which was estimated from the partial pressure in the scattering chamber due to the presence of the molecular beam.
In Section 2.4.5.2., the presence of two distinct adsorption sites on Pt(111) was observed by TPD and helium atom reflectivity measurements. When the activation energy for desorption of carbon monoxide from the two sites was compared to literature reports, these sites were identified as a small population of step-edge sites and a larger population of terrace sites. To account for different rates of methane adsorption at these two sites, two sets of sticking coefficients were determined for each incident translational energy. To calculate the sticking coefficient for adsorption at step-edge sites, the initial rate of adsorption on a fully clean crystal was measured. However, to calculate the sticking coefficient at terrace sites, the rate of adsorption was measured for a partially-covered Pt(111) crystal (0.02 < < 0.05). For adsorption experiments with a Ni(111) crystal, the rate of methane chemisorption across the translational energy range studied was below the threshold of detection (so 10-7).
5.3.1. Adsorption Rate Without Laser Excitation
Both initial and long-term sticking coefficients
for adsorption on Pt(111) are shown as a function of incident
translational energy in Figure 52.
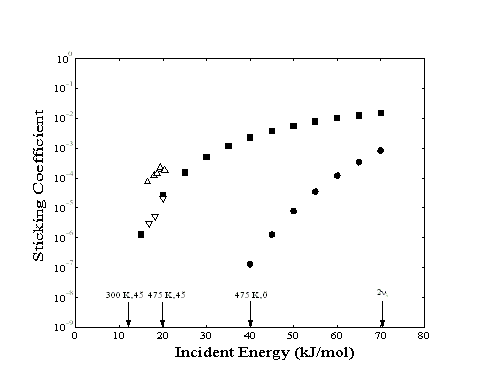
Figure 5-2. Sticking coefficient
of methane on Pt(111) from the initial rate of adsorption (, at
step edges), the long-term rate of adsorption (, at terraces),
and literature reports ()35. The sticking coefficients
of methane on Ni(111) reported by Ceyer are shown for comparison
(). Arrows refer to the translational energy of methane for a
given nozzle temperature and orientation of the sample mount.
The arrow to the extreme right refers to the energy of the 23
vibration.
For comparison, sticking coefficients determined by quantitative Auger electron spectroscopy by Luntz and Bethune35 are also shown. Although the initial sticking coefficients (primarily corresponding to adsorption at step-edges) observed in this study are approximately 10 times higher than the average sticking coefficients reported in the literature, the long-term sticking coefficient (for adsorption at terrace sites) is consistent with the literature values. While the step-edge sites account for less than 2% of all available adsorption sites and do not contribute significantly to the overall average sticking coefficient, the high sensitivity of helium atom reflectivity has made adsorption at these sites observable.
Since the ratio of the two measured sticking coefficients
(sstepedge/sterrace) decreases with increasing
incident translational energy, the greater rate of adsorption
appears to be primarily due to differences in the activation energy
for adsorption at the two sites. At the limit of incident translational
energies greater than the activation energy, the rates of adsorption
at the two sites will converge. In contrast, additional differences
between the two sticking coefficients due to improper calibration
of the cross-section of adsorbates at the two sites would be observed
as a constant factor, independent of the translational energy.
5.3.2. Adsorption Rate With Laser Excitation
For all incident translational energies and nozzle
temperatures, the rate of methane chemisorption on Pt(111) was
independent of laser excitation of the incident methane molecules.
In a sample data set of methane adsorption with initial coverage
equal to =0.03 ML (Figure 5-3), no discontinuity in the rate
of long-term specular decay is observed when the laser radiation
is blocked or subsequently readmitted into the cavity. The initial
rate of adsorption at step-edge sites also does not change due
to laser excitation of the methane molecules (Figure 5-4). However,
the rapid filling of these sites makes the comparison of rates
of specular decay both with and without vibrational excitation
more difficult and less quantitative. Given an average methane
excitation probability of 10% and the 250:1 signal to noise ratio
of a sample data set shown below, this establishes a detectable
upper limit of at most a factor of two enhancement of the adsorption
rate due to vibrational excitation of the impinging methane molecules.
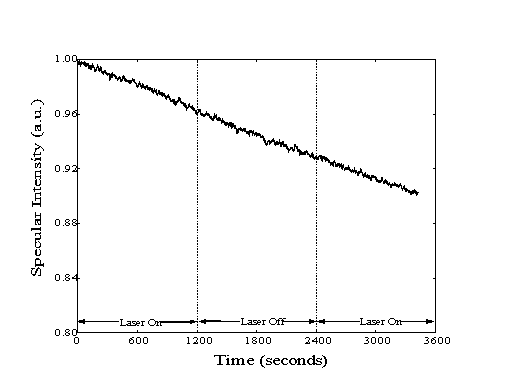
Figure 5-3. Sample helium
specularity measurement during methane adsorption on Pt(111).
Laser light was admitted to the cavity during the first and last
twenty minutes of the experiment. The vibrational excitation
of approximately 10% of all methane molecules in the beam was
confirmed before and after the experiment by bolometric detection.
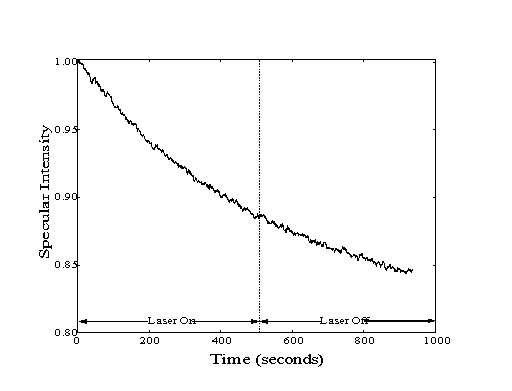
Figure 5-4. Adsorption
on a clean Pt(111) surface shows no discontinuity of specular
decay due to the vibrational excitation of the methane molecules
in the molecular beam. Curvature of the specular decay is due
to reduced vacancy of the step-edge sites.
The rate of adsorption of vibrationally-excited methane on Ni(111) was also explored by helium atom reflectivity studies. However, due to the large activation barrier to methane chemisorption (55 kJ/mol), the sticking coefficient for all experiments was less than the threshold for detection of the experimental apparatus (so=10-7, as estimated from the methane adsorbate scattering cross-section on Pt(111)). Although methane molecules with 71 kJ/mol of translational energy have been observed to chemisorb on Ni(111) with a relatively high initial sticking coefficient (so 10-3)38, an equivalent amount of vibrational energy in the 23 mode was not observed to increase the sticking coefficient above threshold levels. Specular intensity data from a sample methane experiment is shown in Figure 5-5.
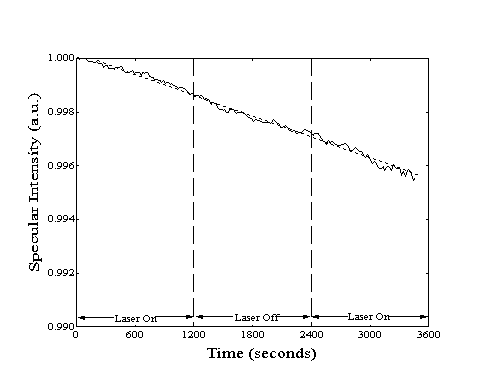
Figure 5-5. Sample helium
specularity measurement during the adsorption of methane on Ni(111).
Note the very different ordinate scale as compared with Figure
5-3. The primary source of the minor specular decay is the adsorption
of background gases. No deviation in the rate of decay is observed
during introduction of the laser to the build-up cavity at the
beginning and end of the experiment.
From this data, it is believed that these preliminary
studies indicate that vibrational energy in the 23
mode is not as effective as translational energy for the promotion
of chemisorption on Ni(111). If vibrational energy in the 3
mode was equally effective as translational energy, the higher
rate of adsorption (s=10-3) of the excited portion
of the beam (10-20%) would have been readily detectable with this
apparatus. This low efficacy of the vibrational energy of the
asymmetric stretching mode for the promotion of
chemisorption is in agreement with the observations of adsorption
on Pt(111). However, due to the inability to measure the low
rate of adsorption of vibrationally inactive methane with this
apparatus, no statements can be made concerning the relative rates
of adsorption on Pt(111) with and without vibrational excitation.
5.3.3. Accommodation of Vibrational Energy
The bolometer was used to detect the internal energy
of the molecular beam both before and after collision with the
surface to determine the vibrational accommodation coefficient
of the clean Pt(111) and Ni(111) surfaces (Figure 5-6). Integration
of the beam profile measurements across all angles was performed
using the techniques outlined in Section 2.4.4.2.
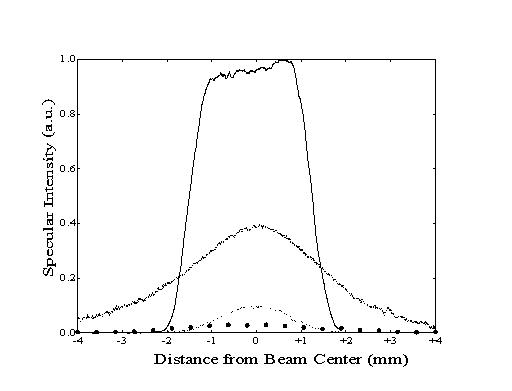
Figure 5-6. Beam profiles generated by bolometric detection of incident beam energy (-), incident vibrational energy (- - -), reflected beam energy (- -), and reflected vibrational energy ( ). Reflected beam data has been magnified for clarity.
Due to differences in the mass of the beam components, the scattering of helium from the Pt(111) surface is more elastic than that of methane. Angular scans (Figure 5-7) were performed by rotating the sample mount through the specular angle (45º) with the mass spectrometer tuned either to m/e=4 (helium) or m/e=15 (methane). For methane, the full-width at half-maximum (FWHM) of the scattering profile is 10º, 2.5 times more than that of helium (4.1º). The scattering of the helium atoms is unaffected by the presence of methane molecules seeded in the beam.
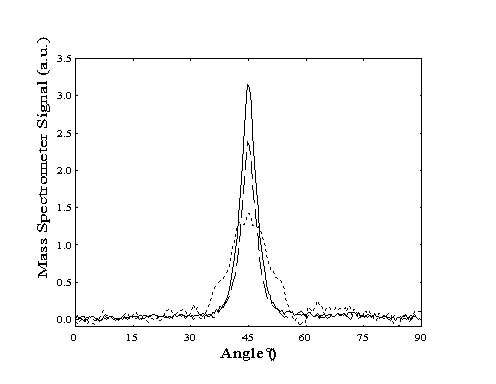
Figure 5-7. Angular scans
of scattered methane (· · ·) and helium (- - -)
from a molecular beam consisting of 2% methane in helium. Scattering
of a pure helium beam (--) is also shown for comparison.
The degree of accommodation can be calculated from the integration of the profile data. For the incident beam, the ratio of the integrated bolometer signals of the total beam energy (Ibeam,inc) and the vibrational energy (Ilaser, inc) is calculated as 13.4. For the reflected beam, the ratio Ibeam,ref / Ilaser,ref is calculated as 15.7, indicating some loss of vibrational excitation between the two bolometer measurements. Using these two ratios and the assumption that all missing vibrational energy was lost exclusively via deactivation upon collision, the upper bound for the survival probability is equal to 0.85.
Although every effort has been made to account for the angular dispersion of the scattered molecules, temporal dispersion (due to loss of translational energy) could not be controlled. Prior to collision with the surface, the modulation of the molecular beam or the laser intensity generates a square pulse with uniform intensity. However, during collision, the broadening of this pulse will be slightly different for the helium and methane in the molecular beam. Since the lock-in amplifier only detects a fraction of the width of the pulse in time, the intensity of the profile data in Figure 5-6 corresponds only to the maximum signal within the pulse.
Alternative loss processes such as spontaneous emission
are unlikely given the short flight time of the beam (34sec) between
the locations of the two bolometer signal measurements as well
as the high energy of the asymmetric stretch overtone. Internal
conversion by intramolecular vibrational energy redistribution
(IVR) or VT transfer does not reduce the energy of the excited
molecule and, as a result, does not reduce the bolometer signal
observed.
5.4. Discussion
Although several molecular beam studies have indicated that the presence of internal energy can increase the rate of direct activated chemisorption of methane on transition metal surfaces4,29, no previous studies have been able to determine if this effect is mode-specific. Modeling of the adsorption data to determine which individual modes are responsible (based on the energy and population of the excited species) have generally been inconclusive. While the models of Winters17 and Beckerle, et al.34 indicated that the excitation of bending modes will increase the sticking coefficient, the quasidiatomic models of Luntz and Holmblad29 indicate that stretching modes potentially are responsible for the observed increases.
At high translational energies, Ceyer and coworkers34 have also proposed that the deformation of the methane molecule upon collision with the surface is able to excite bending vibrations (2, 4). With the duration of the surface encounter much greater than that of a single vibrational period (140 fs vs. 26 fs), any vibrations induced in the molecule could potentially contribute to increased exposure of the carbon atom to the metal surface. Although the majority of vibrational excitation generated by heating the nozzle in molecular beam experiments corresponds to these bending vibrations, there is only slight evidence for vibrational excitation due to collision observed in inelastically scattered methane from Pt(111),
Unfortunately, most literature reported studies
of vibrational energy and adsorption have used diatomic molecules
(e.g. H215 or N228)
with only one available vibrational mode, a symmetric stretch.
Although these studies have clearly demonstrated that stretching
can be effective for the dissociative adsorption of diatomic species
on surfaces, the stretching mode in polyatomics may not be as
useful for increasing reactivity. For methane, increased exposure
of the central carbon atom resulting from the motion of a bending
vibration may be critical for forming a chemical bond with the
surface. While increasing the methyl-hydrogen bond distance by
a stretch may facilitate dissociation of the hydrogen atom, this
may not be the rate limiting step for direct chemisorption.
5.4.2 Accommodation of Vibrational Energy
The observation of a low probability of vibrational deactivation during the methane-surface interaction in Section 5.3.3. is consistent with previous studies of molecular beam-surface collisions. Although deactivation experiments performed with ambient pressures of gases incident on NaCl, molybdenum, silver and platinum surfaces in a gas-cell have shown high deactivation probabilities ranging from 20 to 81%, supersonic beam experiments performed with cleaved LiF(100) or Ag(111) surfaces have shown deactivation rates of less than 10%. For molecular beam experiments, even vibrationally excited large molecules such as butane were observed to suffer minimal vibrational energy loss during collision.
Several mechanisms of vibrational deactivation during collision have been proposed. Electron-hole pair formation, intramolecular energy conversion, or radiative transfer of energy were all feasible processes made possible by long surface residence times encountered during the non-molecular beam experiments. However, in the state-specific study of the collision of NO with graphite surfaces, the primary source of deactivation was identified as interaction with surface phonons. While weak intramolecular coupling between rotational, vibrational, and kinetic energy modes in the molecule prevented self-deactivation, a high degree of rotational accommodation with the surface indicated strong interaction with phonons. This was also confirmed by a surface temperature dependence of the survival probability. At higher surface temperatures, phonons are less able to act as an energy sink, increasing the observed survival of the vibrational excitation.
The interconversion of
some of the vibrational energy from the stretching modes to the
bending modes during the collision of methane with the surface
cannot be ruled out by bolometric measurement (since all vibrational
energy is equivalent to the detector). As a result, the negative
result observed in these experiments may also have some implications
for the bending modes. Potentially, the motion of a highly vibrationally
excited methane molecule is less able to enhance the adsorption
rate than a methane molecule excited in the lower-energy fundamental
modes.
A new experimental apparatus has been developed to determine the role of vibrational energy in the process of methane chemisorption on Ni(111) and Pt(111) surfaces. Using an external-resonant cavity and a color-center laser system, an average excitation of 10% of the methane molecules in the beam was achieved. To measure the rate of adsorption with a high level of sensitivity and precision, helium atom reflectivity was used.
The rate of adsorption of methane on Pt(111) was observed to be independent of excitation of the overtone of the asymmetric stretch (23) for a range of translational energies and methane concentrations. In addition, preliminary experiments indicate that energy of the overtone of the asymmetric stretch is less effective than an equivalent amount of translational energy for the adsorption of methane on Ni(111). From bolometric measurements of the scattered methane, the vibrational energy does not appear to be readily lost to the surface upon collision.
Although reports in the
literature have observed a rate increase attributed to the presence
of vibrationally excited molecules, no reports have indicated
which particular vibrational modes are responsible for the increase.
While these results indicate the 23 overtone is ineffective,
it has yet to be determined if the bending modes (2,
4) or even the fundamental asymmetric stretching mode
(3) may promote the chemisorption of methane.
5.6. Future Experiments
With evidently no increase in dissociative sticking coefficient attributable to excitation of the asymmetric stretching mode, potentially only bending vibrations are able to promote chemisorption. Unfortunately, attempts to find suitable adsorbate/substrate systems with bending vibrations that can be excited by the existing laser system have been unsuccessful. The majority of the molecules with strong vibrational transitions within the available frequency range of the laser system also have high sticking coefficients before vibrational excitation. As a result, any increases in the rate of adsorption would be difficult to resolve. Also, the substrate chosen must be well-ordered and readily cleaned for helium atom reflectivity to be an effective technique for the precise determination of the rate of adsorption.
Combination bands composed of at least one bend vibration were slightly outside the range of the laser for methane and partially deuterated methane (5300-5700 cm-1). Perdeuterated methane transitions at 6280 cm-1 (1 + 3 + 24) and 6520 cm-1 ( 2 + 23 + 4 ) are significantly weaker but potentially detectable. If bending vibrational modes are exclusively responsible for the increase reported in the literature, the sticking coefficient for the excited molecules will change by several orders of magnitude with the addition of 25 kJ/mol of bending vibrational energy (75 kJ/mol total vibrational energy). As a result, the change in sticking coefficient may still be detectable even though the absorption coefficients of these combination bands are 50-200 times weaker than that of the methane vibration explored in the experiments of this chapter. With up to 0.2% of methane in the molecular beam excited, the sticking coefficient would be observed to increase by a detectable 10% if vibrationally excited molecules adsorb only 50 times more readily. Preliminary investigations were unable to locate these transitions due to insufficient availability of precise spectroscopic data.
To explore the possibility that very high vibrational
excitation may actually decrease the probability of chemisorption,
molecules with high sticking coefficients could also be vibrationally
excited. Ethylene, acetylene (1+3, 6529
cm-1), and ammonia (23, 6624 cm-1)
have been observed to readily adsorb to Pt(111) and Ni(111) with
sticking probabilities near unity. However, all three molecules
are observed to adsorb via interactions of electron density
in either -orbitals or "lone-pair" orbitals with the
surface. Although the activation barrier for this process is
readily overcome, the molecule must reside on the surface for
a period of time to orient properly in order to adsorb. If a
vibrationally excited molecule strikes the surface and is less
able to be oriented into a configuration preferable for bonding,
there may be a reduced probability of adsorption.
5.6.1. Laser System Modifications
With the frequency range of the existing build-up cavity and laser combination limited to 5800-6700 cm-1, only a few laser excitation experiments can be performed without significant modification of the current apparatus. To extend the study into additional frequency domains, all three elements must be upgraded: laser, external resonant cavity, and fiber optic cable.
The Scoles lab is currently equipped with a 3.0 m color-center laser which would be useful for exploring the fundamental C-H stretches and bends. However, improvements in diode laser and packaging technology have made moderately-inexpensive "pigtailed" lasers available for several frequencies across the infrared and visible spectrum. Although the power of these lasers (20-45mW) is significantly lower than can be achieved by focusing the output of a tabletop system into a fiber, the high efficiency, compact size and ease of use are strong advantages. However, if the light is directly coupled into the fiber to the cavity, a new system for "locking" the frequency to the transition would need to be developed. Potentially, bolometric detection of a fraction of the excited molecules would be able to provide sufficient feedback without requiring the diversion of photons from the fiber into a scanning etalon or other frequency detection device.
Commercially available external resonant cavities can be purchased from Newport only for frequencies in the visible and near-infrared. However, the principle of design of these devices is relatively simple so it should be possible to manufacture cavities in-house to explore a broader range of frequencies. The response of the piezoelectric tube must be sufficient to change the mirror separation by one free spectral length (calculated as /2). The radii and spacing of the mirrors should be chosen so that the beamwaist is close to the diameter of the molecular beam with considerations made for the preferred size of the device and ease of alignment. Performance as measured by "buildup factor" will be dependent on the reflectance of the mirrors (which can be chosen by the designer) and absorbance (which will be primarily determined by chamber contamination). Although assembly and alignment of the mirrors is critical for performance, experience with the reattachment of mirrors to cavities with faulty epoxy has shown that it is a surprisingly fault-tolerant process.
The last component, the fiber optic cable, is potentially one of the more expensive components to replace. Although fibers for transmission in the visible and near-infrared region (particularly 1.55 m) have been extensively developed and mass produced for the telecommunications industry, fibers suitable for the mid-infrared remain significantly more esoteric and expensive. While the convenience of cabling light across the lab could be sacrificed to save money, the use of the fiber as a feedthrough into the machine ensures high efficiency transfer of light to the cavity and would be difficult to replace with a system of mirrors and lenses.References