3. Physisorption of Hydrocarbons
on Au(111)
3.1. Introduction
The study of the adsorption of hydrocarbons on transition metal surfaces has been a focal point of surface science research for several decades. Using single crystals and precisely controlled deposition conditions, a variety of molecule-surface interactions have been studied. Depending on surface temperature and other experimental parameters which determine the available free energy, hydrocarbons can chemisorb, physisorb, or react with adsorbates already on the surface.
Hindered by the presence of activation barriers and by steric requirements, direct dissociative chemisorption generally cannot readily occur at low surface temperatures. While hydrocarbons are not observed to chemisorb on Au(111) under moderate conditions, investigations of the rate of dissociative adsorption for short alkanes on other more active, lowindex metal surfaces (e.g. Ir(110), Pt(111), Ni(110) and Ni(111)) have shown the activation energy for chemisorption to range from 10 to 50 kJ/mol. To provide enough energy to overcome the barrier to direct chemisorption, either the surface temperature or the translational energy of the incident molecule can be increased. Alternatively, chemisorption can be achieved through a physisorbed precursor-mediated process.
By lowering the surface temperature, a stable coverage of physisorbed molecules can be generated. While in a bound state, each molecule is held in relative proximity to the surface (typically 3.3-3.9 Å for hydrocarbons,) for a period much longer than the few picoseconds of a single elastic or mildly inelastic surface encounter. Although less free energy is available to the molecule at lower surface temperatures to overcome the same chemisorption barrier, the adsorbates are able to make many more attempts at chemisorption before desorbing. Since the chemisorption rate is related to the population of adsorbates and the residence time, the rate of precursor-mediated chemisorption will be seen to decrease when the surface temperature is increased. For large molecules that are still capable of physisorption at high surface temperatures where available free energy is sufficient to overcome the barrier, this precursor-mediated pathway may be highly effective.
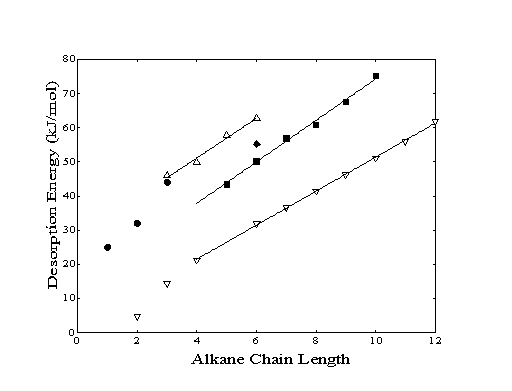
At low surface temperatures (Ts < 200 K for small molecules) and low incident molecular translational energies, hydrocarbons have been observed to physisorb molecularly even on reactive metal surfaces. Low temperature studies of the adsorption of alkanes on Cu(100), Pt(111),, Pt(110), Ir(110), and Ru(001) have measured physisorption energies at partial and full monolayer coverages. Although exact adsorption energies differ slightly due to the atomic structure and the surface potential of the metal substrate, a linear dependence of the adsorption energy on chain length is generally observed (Figure 3-1). The measured energies are proportional to such molecular properties as the polarizability and the bulk enthalpy of vaporization of the adsorbate indicating that dispersion forces are primarily responsible for the physisorption well depth.
While it is found that the incremental bulk heat of vaporization per additional methylene group is 5 kJ/mol for the physisorption of linear alkanes on Cu(100)9 or Ru(001)14, the incremental desorption energy per methylene is 6.5 kJ/mol. A recent study of the adsorption of several hydrocarbons with less than seven carbon atoms on Cu(100) has found that a) the incremental adsorption energy per methylene unit is 6.3 kJ/mol, b) the cyclic hydrocarbons need less energy to evaporate than the corresponding linear alkanes and c) the alkenes need on the order of 4 ± 1 kJ/mol more energy to desorb than the corresponding saturated compounds. Only one study reported to date has quantified the adsorption of alkanes on Au(111), yielding for the physisorption energy of nhexane the value of 55.2 kJ/mol which is similar to the data obtained on other surfaces.
Figure 3-1. The desorption
energies determined by temperature programmed desorption are shown
for alkanes deposited on a variety of surfaces: Cu(100)9
(), Pt(111)10,18
(), Ru(001)14
(), and Au(111)16
(). The enthalpy of vaporization () is also shown for comparison.
As part of a larger study on the self-assembly of
1-alkanethiols on Au(111), the physisorption and chemisorption
of a series of sulfur-containing molecules have been separately
characterized. In the course of that study, it became necessary
to obtain the physisorption energies of several n-alkanes
on Au(111) as a benchmark reference. To complement the linear
alkane data, the study was then expanded to a series of alkenes
and to several cyclic molecules with various structures. Using
the helium atom reflectivity methods described in Chapter 2, the
adsorption energetics and kinetics of these molecules were explored.
The results are reported here together with a bond-additive model
that not only rationalizes the data but also has predictive value.
In addition, the initial sticking coefficients were determined
for the adsorption of the longer-chain, linear alkanes on a Au(111)
crystal, at surface temperatures between 200 K and the desorption
temperature of each hydrocarbon. Sticking coefficients have previously
been reported only for small alkanes on Pt(111),.
These kinetic results are also examined in the light of existing
quantitative models of surface condensation.
3.2. Energies of Adsorption
The measured activation energies for desorption from Au(111) for the linear hydrocarbons studied are summarized in Table 3-1. For the long-chain members (n 6) of both series, there is a linear increase of 6.2 0.2 kJ/mol additional adsorption energy per methylene unit. For the nalkanes, there is a 19.0 1.1 kJ/mol zero offset arising in part from differences between the methyl and methylene subunits. For the 1-alkenes, the zero offset is slightly greater, 20.0 1.3 kJ/mol. When compared to bulk properties such as the heat of vaporization or the average polarizability, the measured energies scale proportionally, pointing to the physical nature of the molecular interaction with the surface.
Although direct comparison between the short-chain
and long-chain alkanes are complicated by the absence of methylene
groups in methane and ethane, the smaller alkanes are, in proportion,
more strongly bound to the surface than their longer-chain counterparts.
The incremental adsorption energy per additional carbon decreases
from 9.6 kJ/mol for ethane to 8.2 kJ/mol for butane to 7.7 kJ/mol
for hexane before reaching the asymptotic value of 6.2 kJ/mol.
For the alkane series, the measured activation energies for desorption are similar to adsorption energies reported in the literature for alkanes deposited on metals other than gold (Figure 3-1). A linear increase of adsorption energy with chain length is indicative that the adsorbate is oriented with the molecular plane (as defined by the backbone of carbon atoms) aligned parallel to the surface. If this alignment was rotated by 90 degrees about the long molecular axis, the methylene groups would be alternatively arranged in high and low positions above the surface. Extending the chain with additional methylenes would give rise to an oddeven chain length effect which would be observed as an alternating series of small and large increases of adsorption energy. Alternatively, placement of the molecule with the long molecular axis normal to the surface would show only a minimal increase of adsorption energy since additional methylene units would be located too far from the surface to contribute to the measured activation energy for desorption.
Desorption experiments with a series of unsaturated
hydrocarbons were performed to explore the dependence of the adsorption
energy on the degree of unsaturation of the molecule as well as
the location of the double bond (Table 3-2). In particular, the
activation energies for desorption of a series of butenes (trans-2-butene,
cis-2-butene, and 2-methylpropene) demonstrated (in agreement
with reference 15) that the location of the double bond within
the molecule does affect the adsorption energy by a few kJ/mol.
For physisorption, the attractive interaction between
the adsorbate and the substrate is due to van der Waals forces.
As the molecule approaches the surface, a correlation arises
between the instantaneous dipole fluctuations present in the metal
and those present in the molecule. As a result, the magnitude
of this attraction can be related to the polarizability of the
molecule. Since polarizability is known to be an additive property,
an empirical energy model was developed to rationalize the data
and to provide predictions of the activation energies for desorption
based on molecular composition.
3.3. Bond Additive Model
3.3.1. Choice of Parameters
Since the total polarizability of hydrocarbons can
be predicted (to within ~1% accuracy) by associating a certain
amount of polarizability to each carbon-hydrogen and carbon-carbon
bond, a model was constructed to determine the contribution of
each bond to the activation energy for desorption observed for
the molecules examined here. To account for the measured values
for the first few members of the alkane and alkene series, it
was necessary to base the contribution of each bond to the adsorption
energy on its location within the molecule (see Figure 3-2 where
heptane, the first hydrocarbon containing an "asymptotic"
methylene group, is shown). Since the incremental methylene contribution
does not reach its asymptotic value before heptane, four carbon
species are required for the model, resulting in four types of
carbon-hydrogen bonds and three types of carbon-carbon bonds (Table
3-3).
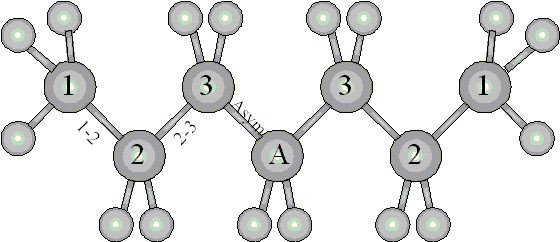
Figure 3-2. Schematic of a heptane molecule showing atom and bond numbering for the additive model. Carbon atoms are numbered towards the core of the molecule. The contribution of all carbons numbered greater than three are considered to be "asymptotic".
Methane, consisting of four carbon-hydrogen bonds, can be used to calculate the adsorption energy contribution of a carbon-hydrogen bond located at the end of the molecule (EType 1 CH = 3.6 kJ/mol). With this value and the activation energy for desorption of ethane, the contribution of a carbon-carbon bond originating from an end carbon atom can also be calculated (EType 1-2 C-C = 24.1 - 6 × 3.6 = 2.5 kJ/mol). For all locations within the molecule, the ratio of the two contributions (ECC / EC-H = 1.44) will be assumed to be constant.
Using this ratio, the asymptotic methylene contribution of 6.2 kJ/mol can be partitioned between two carbon-hydrogen bonds (2.3 kJ/mol each) and one carbon-carbon bond (1.6 kJ/mol). For simplicity, the contributions of the intermediate carbon-hydrogen and carboncarbon bonds have been linearly interpolated from the methyl and methylene extreme values (Figure 3-2). To reflect the intermediate nature of bonds located in cyclic molecules, the activation energy for desorption can be calculated as the sum of the contributions of intermediate bonds (Type 2 C-H bonds and Type 2-3 C-C bonds).
Using these additive bond contributions and the
experimental activation energy for the desorption of 1-octene,
the double bond contribution can be derived to be 10.5 kJ/mol.
Although this model will not distinguish between double bonds
with differing positions within the molecule, its performance
for both saturated and unsaturated hydrocarbons is quite satisfactory
and will be discussed in the following section.
3.3.2. Model Performance
The comparison between the observed and calculated
activation energies for desorption is presented in Table 3-4.
Since the energy of physisorption depends on an additive property,
i. e. the polarizability, it is not surprising that
this additive model predicts the observed activation energy for
desorption relatively well. The model, which contains four free
parameters, accounts for the experimental energies of 25 saturated
and unsaturated
| |||
hydrocarbons with an average error of 1.9%. Of these
25 molecules, 84% of the measured adsorption energies differ from
the calculated values by less than twice the average error. It
is the deviation of the model predictions from experimental values
that is likely to provide the most useful information. The greatest
error occurs for ethylene and 1,3-butadiene, with underestimation
of the activation energy for desorption by 58%. The origin
of this error appears to be related to the higher binding energy
contribution of the end groups in the saturated chains. Having
determined the contribution of the double bond from a long-chain
1alkene (a molecule with both an unsaturated region and
a large saturated region) some deviation may be expected for the
smaller alkenes. It is possible that this end effect deviation
may result from the deformation of the charge density of the metal
surface around the adsorbed molecule which would have a larger
effect on the ends of the molecule than on the center. This view
is confirmed by the fact that good agreement between experiment
and model is achieved for cyclohexane and benzene but not for
cyclooctane. To obtain better agreement for this molecule, the
contribution of the carbon-hydrogen and carbon-carbon bonds should
be smaller, i.e. closer to the asymptotic value. The trend
is, therefore explainable since the increased size of the ring
in the limit of a very large cycloalkane would create an environment
similar to that of a long linear alkane.
3.3.3. Extension to Sulfur-Containing Molecules
In order to test the model on a wider range of molecules, the data on the activation energy for desorption of alkanethiols and other sulfur-containing molecules which were collected on this apparatus and reported in Chapter 4 and an earlier paper18 were reexamined. The value for the sulfur atom contribution was determined from the difference in the y-offsets of the alkane and alkanethiol series as 24.1 kJ/mol. For simplicity, this value was also assumed to be constant for all molecules. The contribution of the sulfur-hydrogen bond was determined to be 8.1 kJ/mol from the alkanethiol data as the average difference between each observed activation energy for desorption and the sum of the contribution of the known components (carbon-carbon bonds, carbon-hydrogen bonds, and sulfur atoms of a particular molecule). For the present purpose, the carbon atoms have been numbered as if the sulfur was a carbon atom.
The agreement between the experimental and calculated adsorption energies for the sulfur-containing molecules is somewhat worse than that for the hydrocarbons. While this could be expected because of the simplicity of the model, the calculated adsorption energies of both ethanethiol and tetradecanethiol deviate sufficiently from the experimental data to warrant a closer look.
For ethanethiol, the model severely underpredicts the activation energy for desorption (8.6%). As ethanethiol is the shortest alkanethiol studied, potentially this deviation is due to the same "end effect" as observed with the alkanes and alkenes. However, the even larger underprediction of tetradecanethiol (13%) is likely to be due to an entirely different cause
(i.e. experimental error). For long chain-lengths
the incremental energy per additional methylene group (as found
in the hydrocarbon series) is asymptotically constant. As a result,
the experimental activation energy for desorption of tetradecanethiol,
which lies well above the linear best-fit that represents most
of the other alkanethiols, is very likely to be incorrect (i.e.
too high).
3.4. Sticking Coefficients
At low temperatures, the initial sticking coefficient (so) is near unity for all the hydrocarbons tested. However, a decrease in sticking coefficient is observed as the surface temperature is increased. This effect does not become pronounced until the surface temperature is approximately 50 K below the temperature corresponding to the observed TPD desorption peak of each species. At surface temperatures above the characteristic TPD temperature, the sticking coefficient approaches zero (Figure 3-3).
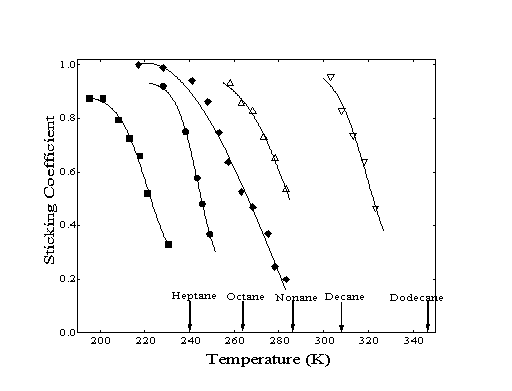
Figure 3-3. The sticking
coefficients as a function of absolute temperature are shown for
the n-alkanes: heptane (), octane (), nonane (), decane
(), dodecane (). Arrows correspond to the peak desorption temperature
determined during temperature programmed desorption measurements
for each molecule.
To compare the sticking behavior of different molecules, the actual surface temperatures were converted to a reduced temperature, T*. This reduced temperature is calculated as the ratio of the actual surface temperature to the peak desorption temperature as observed by TPD for each species. When the sticking coefficients are replotted as a function of the reduced temperature, the sticking coefficient curves overlap for all the n-alkanes and 1alkenes (Figure 3-4) indicating that the mechanism of adsorption is independent of the identity of the adsorbate. Similar results have also been observed using the same apparatus for the physisorption of 1-alkanethiols comprised of two to ten carbons.
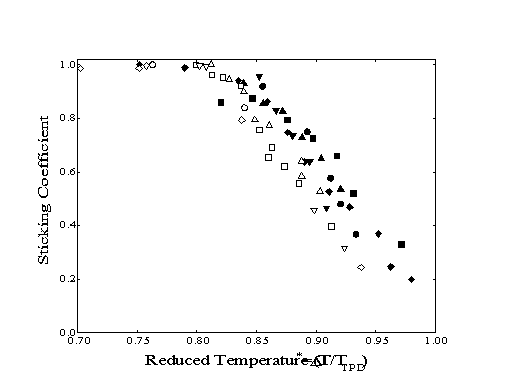
Figure 3-4. The sticking
coefficient of n-alkanes (filled) and 1-alkenes (open)
as a function of reduced temperature relative to the TPD peak
for each molecule.
A similar sticking coefficient dependence on surface temperature was observed by Arumainayagam et al.11 in their studies of the adsorption of ethane on Pt(111). At surface temperatures close to the desorption temperature, trapping probabilities as measured by the technique of King and Wells were observed to decrease significantly. According to hard-cube models, trapping probability should have had little dependence on surface temperature so the differences were attributed to desorption within the experimental time resolution of one second. To quantify the desorption effects, a model was generated to predict the apparent trapping coefficient for a system where the true sticking coefficient was unity, independent of temperature. Although the model successfully accounted for the general shape of the trapping coefficient curve as a function of surface temperature (shown in Figure 9 of reference 11), a deviation from observed data still remained which was significant only at surface temperatures close to the desorption temperature (across the range T*=0.75 to 1.0).
An estimate of the sticking coefficient as a function of surface temperature can be generated from the data and model of Arumainayagam et al.11 by calculating the ratio of the observed trapping probabilities divided by the predicted trapping probabilities. Since the model accounts for the reduction in the apparent trapping probability due to desorption, any deviation from the predicted values will be real. For low surface temperatures, there is no deviation between the observed data and the model, and a sticking coefficient of one can be calculated. However, at surface temperatures closer to the peak desorption temperature (160 K), the observed and predicted trapping probabilities diverge. When the estimated sticking coefficients generated in this way are plotted as a function of reduced temperature, the resulting plot appears similar to the long chain alkane sticking coefficient data determined in this study.
Due to the qualitatively similar appearance of the effects of desorption and the observed dependence of the sticking coefficient on surface temperature, it was necessary to determine whether sticking coefficients or condensation coefficients were being measured using the techniques outlined in Section 2.3. Whereas the condensation coefficient is dependent on the process of desorption (because it measures the net change in adsorbate population per unit flux), the sticking coefficient measures only the probability of adsorption independently from the flux. As a result, the sticking coefficient will be independent of both the impingement rate and the rate of desorption.
In an extensive study of the adsorption of water on ice multilayers, Kay and collaborators21 observed the dependence of the condensation coefficient on surface temperature and flux while the sticking coefficient remained unity. By increasing the surface temperature (and consequently, the rate of desorption), the condensation coefficient decreased from unity to zero. However, as flux was increased, the measured condensation coefficient also increased for a given surface temperature. By comparison, examination of sticking coefficient data collected for 1-hexene across approximately two decades of flux shows no sticking coefficient dependence on flux (Figure 3-5). This provides evidence that the true sticking coefficients are being reported here.
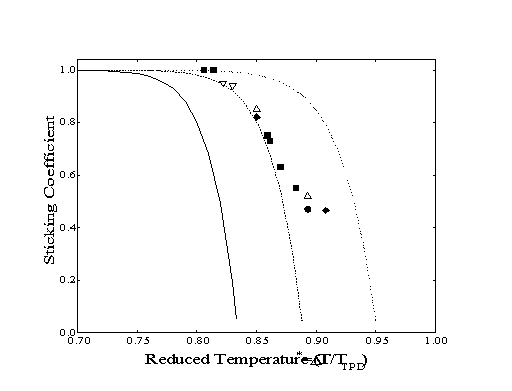
Figure 3-5. The sticking coefficient of 1-hexene as a function of reduced temperature for a series of approximate dosing rates: 510-4 ML/sec (), 210-3 ML/sec (), 510-3 ML/sec (), 8103 ML/sec (), 510-2 ML/sec (). Condensation coefficients were also calculated using the model of Brown, et al.21 modified for first-order desorption and a temperature-independent sticking coefficient equal to one: 5104 ML/sec (--), 510-3 ML/sec (- -), 510-2 ML/sec (· · · ·).
Using the data analysis procedure presented in this paper, no additional modeling is required to modify or normalize the observed sticking coefficients. With the technique of helium atom scattering, surface populations are measured directly during dosing so there is no dead-time before measurements when desorption may reduce the actual quantity of adsorbates. Any effects of desorption on coverage have already been fully accounted for by the Langmuir adsorption model used to derive the sticking coefficient values. If desorption rates were systematically estimated incorrectly (due to miscalibration of the surface temperature thermocouple for example), sticking coefficients measured at surface temperatures where desorption is significant would depend on the incoming flux. Similarly, if impingement rates are inaccurate, measurements of sticking at low surface temperatures would deviate from unity.
The origin of the decrease in sticking coefficient
therefore appears to be due to incomplete accommodation of the
molecule by the Au(111) surface at higher temperatures. Even
though the adsorbate molecules are only arriving with an average
of 3.6 kJ/mol of translational energy (from E =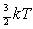 at 300 K), this quantity is sufficient to allow the molecule to
avoid physisorption at surface temperatures near the peak desorption
temperature. At low temperature, both phonon creation and intramolecular
energy transfer from translational to rotational (and less likely,
vibrational modes) can allow the molecule to become trapped in
the physisorption well of the surface. However, at higher surface
temperatures, the surface is unable to readily accept energy from
the molecule resulting in incomplete accommodation.
at 300 K), this quantity is sufficient to allow the molecule to
avoid physisorption at surface temperatures near the peak desorption
temperature. At low temperature, both phonon creation and intramolecular
energy transfer from translational to rotational (and less likely,
vibrational modes) can allow the molecule to become trapped in
the physisorption well of the surface. However, at higher surface
temperatures, the surface is unable to readily accept energy from
the molecule resulting in incomplete accommodation.
From this set of results, it is unclear whether
the procedure of dosing from ambient background gas instead of
from a monoenergetic molecular beam has affected the values of
the reported sticking coefficients. For ethane, the sticking
coefficients derived from the results of Arumainayagam et al.11
show no dependence on translational energy between 10 and 24 kJ/mol.
Therefore, it is unlikely that the observed decrease in sticking
coefficient at higher surface temperatures is a result of increasingly
selective physisorption of less translationally energetic molecules.
3.5. Summary
The kinetics and energetics of the physisorption of thermal energy n-alkanes and 1alkenes on Au(111) have been determined. For both systems, adsorption on the surface proceeds through a physisorbed state and can be modeled using Langmuir first-order adsorption kinetics. At higher surface temperatures, the physisorption sticking probability decreases due to incomplete accommodation by the surface for both hydrocarbon series.
For the long chain-hydrocarbons, the contribution of each additional methylene group to the activation energy for desorption from a physisorbed state is 6.2 0.2 kJ/mol. All molecules studied, both linear and cyclic bind with the molecular plane oriented parallel to the surface. A bond-additive empirical model has been presented which is able to predict the observed physisorption energies for 25 hydrocarbons with an average error of less than 2.0%. If sulfursubstituted hydrocarbons are also considered, the average error increases to 2.6%
(for 35 molecules).
The physisorption sticking coefficient has been established for straight chain alkanes and alkenes across a range of temperatures. For molecules incident with thermal kinetic energy (as generated by dosing through a leak valve), the initial sticking coefficient is near unity at low temperatures. This value decreases as the surface temperature is raised closer to the temperature corresponding to the observed TPD desorption peak. Even after a higher rate of desorption is accounted for, a decrease in sticking can be seen. This temperature dependent behavior is similar for all linear molecules studied.
References
(1) See references in Somorjai, G. A. Chemistry in Two Dimensions: Surfaces; Cornell University Press: Ithaca, NY, 1981. and Somorjai, G. A. Introduction to Surface Chemistry and Catalysis; John Wiley and Sons: New York, 1994.
(2) Chesters, M. A.; Somorjai, G. A. Surf. Sci. 1975, 52, 21.
(3) Hamza, A. V.; Steinruck, H. P.; Madix, R. J. J. Chem. Phys. 1987, 86, 6506.
(4) Schoofs, G. R.; Arumainayagam, C. R.; McMaster, M. C.; Madix, R. Surf. Sci. 1989, 215, 1.
(5) Ceyer, S. T. Ann. Rev. Phys. Chem. 1988, 39, 479 and references therein.
(6) Balasubramanian, S.; Klein, M. L.; Siepmann, J. I. J. Phys. Chem. 1996, 100, 11960.
(7) Gupta, S.; Koopman, D. C.; Westermann-Clark, G. B.; Bitsanis, I. A. J. Chem. Phys. 1994, 100, 8444.
(8) Sullivan, D. J. D.; Flaum, H. C.; Kummel, A. C. J. Phys. Chem. 1993, 97, 12051.
(9) Sexton, B. A.; Hughes, A. E. Surf. Sci. 1984, 140, 227.
(10) McMaster, M. C.; Arumainayagam, C. R.; Madix, R. J. Chem. Phys. 1993, 177, 461.
(11) Arumainayagam, C. R.; Schoofs, G. R.; McMaster, M. C.; Madix, R. J. J. Phys. Chem. 1991, 95, 1041.
(12) McMaster, M. C.; Schroeder, S. L. M.; Madix, R. J. Surf. Sci. 1993, 297, 253.
(13) Kang, H. C.; Mullins, C. B.; Weinberg, W. H. J. Chem Phys. 1990, 92, 1397.
(14) Brand, J. L.; Arena, M. V.; Deckert, A. A.; George, S. M. J. Chem. Phys. 1990, 92, 5136.
(15) Teplyakov, A. V.; Gurevich, A. B.; Yang, M. X.; Bent, B. E.; Chen, J. G. Surf. Sci. 1998, 396, 340.
(16) Dubois, L. H.; Zegarski, B. R.; Nuzzo, R. G. J. Am. Chem. Soc. 1990, 112, 570.
(17) Majer, V.; Svoboda, V. Enthalpies of Vaporization of Organic Compounds; Blackwell Scientific Publications; Boston, MA, 1985. 73-86.
(18) Lavrich, D. J.; Wetterer, S. M.; Bernasek, S. L.; Scoles, G. J. Phys. Chem. B 1998, 102, 3456.
(19) Arumainayagam, C. R.; McMaster, M. C.; Schoofs, G. R. Surf. Sci. 1989, 222, 213.
(20) Stinnett, J. A.; Madix, R. J. J. Chem. Phys. 1996, 105, 1609.
(21) Brown, D. E.; George, S. M.; Huang, C.; Wong, K. L.; Rider, K. B.; Smith, R. S.; Kay, B. D. J. Phys. Chem. 1996, 100, 4988.
(22) Le Fèvre, C. G.; Le Fèvre, R. J. W.; Rao, B. P.; Smith, M. R. J. Chem. Soc. 1959, 1188.
(23) We calculated the number of parameters chosen to optimize the fit (4) as follows: 1) the terminal C-H contribution from methane, 2) the terminal C-C contribution from ethane, 3) the asymptotic C-H and C-C contribution from the slope of best linear fit of the activation energy for desorption versus chain length, and 4) the C=C contribution from 1-octene. The other C-H and C-C bond contributions are interpolated and therefore are not considered as "free" parameters.
(24) Levi, A.; Scoles, G., unpublished work.
(25) The results of this chapter were submitted for publication to J. Phys. Chem. B.