SELECTED PAPERS
Click on the images to access full-text publications.
Cytoskeleton 2010; 67(9): 599-607.
Tropomyosin is a tetramer under physiological salt conditions.
Lassing, I, Hillberg, L, Hoglund, A-S, Karlsson, R, Schutt, CE, Lindberg, U
Tropomyosin (TM) is a coiled-coil dimer of a-helical peptides, which self associates in a head- to-tail fashion along
actin polymers, conferring stability to the microfilaments and serving a regulatory function in acto-myosin driven
force generation. While the major amount of TM is associated with filaments also in non-muscle cells, it was recently
reported that there are isoform-specific pools of TM multimers (not associated with F-actin), which appear to be utilized
during actin polymerization and reformed during depolymerization. To determine the size of these multimers,
skeletal muscle TM was studied under different salt conditions using gel-filtration and sucrose gradient sedimentation,
and compared with purified non-muscle TM 1 and 5, as well as with TM present in non-muscle cell extracts and skeletal muscle TM added to such extracts. Under physiological salt conditions TM appears as a single homogenous peak with the Stokes radius 8.2 nm and the molecular weight (mw) 130,000. The corresponding values for TM 5 are 7.7 nm and 104,000, respectively. This equals four peptides, implying that native TM is a tetramer in physiological salt. It is therefore concluded that the TM multimers are tetramers.
|
Click on image below to
access full-text publication...
|
Seminars in Cancer Biology 2008; 18: 2-11.
The Microfilament System and Malignancy.
Lindberg, U, Karlsson, R, Lassing, I, Schutt, CE, Hoglund, A-S
Increased motile activity, increased rate of cell proliferation and removal of growth inhibiting cell–cell contacts are hallmarks of tumorigenesis. Activation of cell motility and migration is caused by activation of receptors, turning on the growth cycle. Increased expression of metalloproteinases, breaking cell:cell contacts and organ confines, allows the spread of malignant cancer cells to other sites in the organism. It has become increasingly clear that most transmembrane proteins (growth factor receptors, adhesion proteins and ion channels) are either permanently or transiently associated with the sub-membraneous system of actin microfilaments (MF), whose force generating capacity they control. Although there has been great progress in our understanding of the physiological importance of the MF-system, as will be exemplified in this issue of SCB, many aspects of actin microfilament formation and its regulation are still unclear. Redox control of the actin (MF)-system in cell motility and migration and its perturbations in pathophysiology, including cancer, is an emerging field of research. |
Click on image below to
access full-text publication..
|
J. of Appl. Cryst. 2008; 41: 600-605.
Protein Crystals can be Incommensurately Modulated.
Lovelace, JJ, Murphy, CR, Daniels, L, Narayan, K, Schutt, CE, Lindberg, U, Svensson, C, Borgstahl, GEO
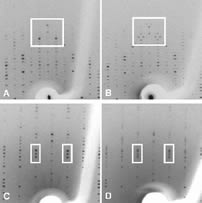
For a normal periodic crystal, the X-ray diffraction pattern can be described by an orientation matrix and a set of three integers that indicate the reciprocal lattice points. Those integers determine the spacing along the reciprocal lattice directions. In aperiodic crystals, the diffraction pattern is modulated and the standard periodic main reflections are surrounded by satellite reflections. The successful indexing and refinement of the main unit cell and q vector using TWINSOLVE, developed by Svensson [(2003). Lund University, Sweden], are reported here for an incommensurately modulated, aperiodic crystal of a profilin:actin complex. The indexing showed that the modulation is along the b direction in the crystal, which corresponds to an ‘actin ribbon’ formed by the crystal lattice. Interestingly, the transition to the aperiodic state was shown to be reversible and the diffraction pattern returned to the periodic state during data collection. It is likely that the protein underwent a conformational change that affected the neighbouring profilin:actin molecules in such a way as to produce the observed modulation in the diffraction pattern. Future work will aim to trap the incommensurately modulated crystal state, for example using cryocooling or chemical crosslinking, thus allowing complete X-ray data to be collected. |
Click on image below to
access full-text publication.
 |
|
J. of Mol. Bio . 2007; 370: 331-348.
Molecular and Structural Basis for Redox Regulation of β-actin.
Lassing, I, Schmitzberger, F, Bjornstedt, M, Holmgren, A, Nordlund, P, Schutt, C, Lindberg, U
An essential consequence of growth factor-mediated signal transduction is the generation of intracellular H2O2. It operates as a second messenger in the control of actin microfilament dynamics, causing rapid and dramatic changes in the morphology and motile activity of stimulated cells. Little is understood about the molecular mechanisms causing these changes in the actin system. Here, it is shown that H2O2 acts directly upon several levels of this system, and some of the mechanistic effects are detailed. We describe the impact of oxidation on the polymerizability of non-muscle β/γ-actin and compare with that of muscle α-actin. Oxidation of β/γ-actin can cause a complete loss of polymerizability, crucially, reversible by the thioredoxin system. Further, oxidation of the actin impedes its interaction with profilin and causes depolymerization of filamentous actin. The effects of oxidation are critically dependent on the nucleotide state and the concentration of Ca2+. We have determined the crystal structure of oxidized β-actin to a resolution of 2.6 Å. The arrangement in the crystal implies an antiparallel homodimer connected by an intermolecular disulfide bond involving cysteine 374. Our data indicate that this dimer forms under non-polymerizing and oxidizing conditions. We identify oxidation of cysteine 272 in the crystallized actin dimer, likely to a cysteine sulfinic acid. In β/γ-actin, this is the cysteine residue most reactive towards H2O2 in solution, and we suggest plausible structural determinants for its reactivity. No other oxidative modification was obvious in the structure, highlighting the specificity of the oxidation by H2O2. Possible consequences of the observed effects in a cellular context and their potential relevance are discussed. |
Click on image below to
access full-text publication.
|
Eur. J. Cell Biol. 2006; 85: 399-409.
Tropomyosins are present in lamellipodia of motile cells.
Hillberg, L, Rathje, L-SZ Nyakern-Meazza, M, Helfand, B, Goldman, RD, Schutt, CE, Lindberg, U.
This paper shows that high-molecular-weight tropomyosins (TMs), as well as shorter isoforms of this protein, are present in significant amounts in lamellipodia and filopodia of spreading normal and transformed cells. The presence of TM in these locales was ascertained by staining of cells with antibodies reacting with endogenous TMs and through the expression of hemaglutinin- and green fluorescent protein-tagged TM isoforms. The observations are contrary to recent reports suggesting the absence of TMs in regions,where polymerization of actin takes place, and indicate that the view of the role of TM in the formation of actin filaments needs to be significantly revised. |
Click on image below to
access full-text publication.
 |
| Advances in Molecular and Cell Biology 2006; 37: 49-66.
The Connection Between Actin ATPase and Polymerization.
Schuler, H, Karlson, R, Schutt, CE, and Lindberg, U.
Remodeling of the actin filament system in cells results from strictly regulated polymerization and depolymerization of actin, where hydrolysis of actin-bound ATP is crucial. Actin–actin interactions are influenced by the state of the bound nucleotide, and many microfilament regulators influence the actin ATPase by binding preferentially either to ATP/ADP.Pi‐ or ADP‐bound actin. This chapter summarizes observations made concerning the actin ATPase and its role in the biological activity of actin and actin filaments. |
Click on image below to
access full-text publication.
|
J. of Appl. Cryst. 2004; 37: 327-330.
Imaging Modulated Reflections From a Semi-Crystalline State of Profilin: Actin Crystals.
Lovelace JJ, Narayan K, Chik JK, Bellamy HD, Snell EH, Lindberg U, Schutt CE, Borgstahl GEO
Modulated protein crystals remain terra incognita for most crystallographers. While small-molecule crystallographers have successfully wrestled with and conquered this type of structure determination, to date no modulated macromolecular structures have been reported. Profilin: β-actin in a modulated semi-crystalline state presents a challenge of sufficient biological significance to motivate the development of methods for the accurate collection of data on the complex diffraction pattern and, ultimately, the solution of its structure. In the present work, fine phi-sliced data collection was used to resolve the closely spaced satellite reflections from these polymorphic crystals. Image-processing methods were used to visualize these data for comparison with the original precession data. These preliminary data demonstrate the feasibility of using fine phi-slicing to collect accurately the intensities and positions of the main and satellite reflections from these modulated protein crystals. |
Click on image below to
access full-text publication.
 |
FEBS Lett. 2003; 552 (2-3): 82-85.
Activation in Isolation: Exposure of the Actin-Binding Site in the C-terminal Half of Gelsolin Does Not Require Actin.
Narayan K, Chumnarnsilpa S, Choe H, Irobi E, Urosev D, Lindberg U, Schutt CE, Burtnick LD, Robinson RC
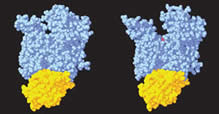
Gelsolin requires activation to carry out its severing and capping activities on F-actin. Here, we present the structure of the isolated C-terminal half of gelsolin (G4-G6) at 2.0 Å resolution in the presence of Ca2+ ions. This structure completes a triptych of the states of activation of G4-G6 that illuminates its role in the function of gelsolin. Activated G4-G6 displays an open conformation, with the actin-binding site on G4 fully exposed and all three type-2 Ca2+ sites occupied. Neither actin nor the type-l Ca2+, which normally is sandwiched between actin and G4, is required to achieve this conformation.
|
Click on image below to
access full-text publication.
 |
J. Biol. Chem. 2002; 277 (32): 28774-28779.
Tropomyosin and Gelsolin Cooperate in Controlling the Microfilament System.
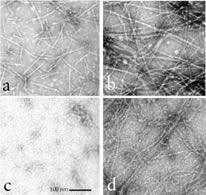
Nyakern-Meazza M, Narayan K, Schutt CE, Lindberg U
Tropomyosin has been shown to cause annealing of gelsolin-capped actin filaments. Here we show that tropomyosin is highly efficient in transforming even the smallest gelsolin-actin complexes into long actin filaments. At low concentrations of tropomyosin, the effect of tropomyosin depends on the length of the actin oligomer, and the cooperative nature of the process is a direct indication that tropomyosin induces a conformational change in the gelsolin-actin complexes, altering the structure at the actin (+) end such that capping by gelsolin is abolished. At increased concentrations of tropomyosin, heterodimers, trimers, and tetramers are converted to actin filaments. In addition, evidence is presented demonstrating that gelsolin, once removed from the (+) end of the actin, can reassociate with the newly formed tropomyosin-decorated actin filaments. Interestingly, the binding of gelsolin to the tropomyosin-actin filament complexes saturates at 2 gelsolin molecules per 14 actin and 2 tropomyosins, i.e. two gelsolins per tropomyosin-regulatory unit along the filament. These observations support the view that both tropomyosin and gelsolin are likely to have important functions in addition to those proposed earlier. |
Click on image below to
access full-text publication.
 |
J. Mol. Biol. 2002; 317 (4): 577-589.
The Role of MeH73 in Actin Polymerization and ATP Hydrolysis.
Nyman T, Schuler H, Korenbaum E, Schutt CE, Karlsson R, Lindberg U
In actin from many species H73 is methylated, but the function of this rare post-translational modification is unknown. Although not within bonding distance, it is located close to the gamma-phosphate of the actin-bound ATP. In most crystal structures of actin, the delta1-nitrogen of the methylated H73 forms a hydrogen bond with the carbonyl of G158. This hydrogen bond spans the gap separating subdomains 2 and 4, thereby contributing to the forces that close the interdomain cleft around the ATP polyphosphate tail. A second hydrogen bond stabilizing interdomain closure exists between R183 and Y69. In the closed-to-open transition in beta-actin, both of these hydrogen bonds are broken as the phosphate tail is exposed to solvent.Here we describe the isolation and characterization of a mutant beta-actin (H73A) expressed in the yeast Saccharomyces cerevisiae. The properties of the mutant are compared to those of wild-type beta-actin, also expressed in yeast. Yeast does not have the methyl transferase necessary to methylate recombinant beta-actin. Thus, the polymerization properties of yeast-expressed wild-type beta-actin can be compared with normally methylated beta-actin isolated from calf thymus. Since earlier studies of the actin ATPase almost invariably employed rabbit skeletal alpha-actin, this isoform was included in these comparative studies on the polymerization, ATP hydrolysis, and phosphate release of actin.It was found that H73A-actin exchanged ATP at an increased rate, and was less stable than yeast-expressed wild-type actin, indicating that the mutation affects the spatial relationship between the two domains of actin which embrace the nucleotide. At physiological concentrations of Mg2+, the kinetics of ATP hydrolysis of the mutant actin were unaffected, but polymer formation was delayed. The comparison of methylated and unmethylated beta-actin revealed that in the absence of a methyl group on H73, ATP hydrolysis and phosphate release occurred prior to, and seemingly independently of, filament formation. The comparison of beta and alpha-actin revealed differences in the timing and relative rates of ATP hydrolysis and P(i)-release.
|
Click on image below to
access full-text publication.
 |
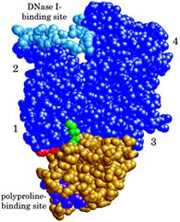
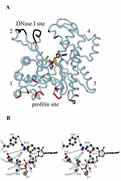
J. Biol. Chem. 2002; 277 (18): 15828-15833.
A Cross-Linked Profilin-Actin Heterodimer Interferes With Elongation at the Fast-Growing End of F-Actin.
Nyman T, Page R, Schutt CE, Karlsson R, Lindberg U
Profilin and b/g-actin from calf thymus were covalently linked using the zero-length cross-linker 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide in combination with N-hydroxysuccinimide, yielding a single product with an apparent molecular mass of 60 kDa. Sequence analysis and x-ray crystallographic investigations showed that the cross-linked residues were glutamic acid 82 of profilin and lysine 113 of actin. The cross-linked complex was shown to bind with high affinity to deoxyribonuclease I and poly(L-proline). It also bound and exchanged ATP with kinetics close to that of unmodified profilin-actin and inhibited the intrinsic ATPase activity of actin. This inhibition occurred even in conditions where actin normally forms filaments. By these criteria the cross-linked profilin-actin complex retains the characteristics of unmodified profilin-actin. However, the cross-linked complex did not form filaments nor copolymerized with unmodified actin, but did interfere with elongation of actin filaments in a concentration-dependent manner. These results support a polymerization mechanism where the profilin-actin heterodimer binds to the (+)-end of actin filaments, followed by dissociation of profilin, and ATP hydrolysis and P(i) release from the actin subunit as it assumes its stable conformation in the helical filament. |
Click on image below to
access full-text publication.
 |
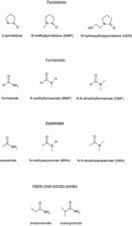
Nucl. Acids Res. 2001; 29 (11): 2377-81.
The Enhancement of PCR Amplification by Low Molecular Weight Amides.
Chakrabarti R, Schutt CE
Amplification of a DNA target by the polymerase chain reaction (PCR) often requires laborious optimization efforts. In this regard, the use of certain organic chemicals such as dimethyl sulfoxide, polyethylene glycol, betaine and formamide as cosolvents has been found to be very helpful. Unfortunately, very little is known about the precise structural features that make these additives effective and, accordingly, the number of such chemicals currently known to enhance PCR is limited. In order to address these issues, we decided to focus on formamide and undertook an extensive study of low molecular weight amides as a class to see how changing the substituents in the amide structure influences its effect on PCR. We describe here the results of this study, which involved 11 different amides, and present observations that provide a cohesive picture of structure-activity relations in this group of additives. We found several of these amides to be exceptionally effective and introduce them as novel PCR enhancers.
|
Click on image below to
access full-text publication.
 |
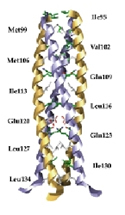
J. Mol. Biol. 2000; 304 (5):861-871.
Crystal Structure of the Oligomerization Domain of NSP4 from Rotavirus Reveals a Core Metal-Binding Site.
Bowman GD, Nodelman IM, Levy O, Lin SL, Tian P, Zamb TJ, Udem SA, Venkataraghavan B, Schutt CE
During the maturation of rotaviral particles, non-structural protein 4 (NSP4) plays a critical role in the translocation of the immature capsid into the lumen of the endoplasmic reticulum. Full-length NSP4 and a 22 amino acid peptide (NSP4(114-135)) derived from this protein have been shown to induce diarrhea in young mice in an age-dependent manner, and may therefore be the agent responsible for rotavirally-induced symptoms. We have determined the crystal structure of the oligomerization domain of NSP4 which spans residues 95 to 137 (NSP4(95-137)). NSP4(95-137) self-associates into a parallel, tetrameric coiled-coil, with the hydrophobic core interrupted by three polar layers occupying a and d-heptad positions. Side-chains from two consecutive polar layers, consisting of four Gln123 and two of the four Glu120 residues, coordinate a divalent cation. Two independent structures built from MAD-phased data indicated the presence of a strontium and calcium ion bound at this site, respectively. This metal-binding site appears to play an important role in stabilizing the homo-tetramer, which has implications for the engagement of NSP4 as an enterotoxin. |
Click on image below to
access full-text publication.
|
Anat. Rec. (New Anat.) 2000; 261:198-216.
The New Architectonics: An Invitation To Structural Biology.
Schutt CE and Lindberg U
The philosophy of art might offer an epistemological basis for talking about the complexity of biological molecules in a meaningful way. The analysis of artistic compositions requires the resolution of intrinsic tensions between disparate sensory categories-color, line and form-not unlike those encountered in looking at the surfaces of protein molecules, where charge, polarity, hydrophobicity, and shape compete for our attentions. Complex living systems exhibit behaviors such as contraction waves moving along muscle fibers, or shivers passing through the growth cones of migrating neurons, that are easy to describe with common words, but difficult to explain in terms of the language of chemistry. The problem follows from a lack of everyday experience with processes that move towards equilibrium by switching between crystalline order and chain-like disorder, a commonplace occurrence in the submicroscopic world of proteins. Since most of what is understood about protein function comes from studies of isolated macromolecules in solution, a serious gap exists between what we know and what we would like to know about organized biological systems. Closing this gap can be achieved by recognizing that protein molecules reside in gradients of Gibbs free energy, where local forces and movements can be large compared with Brownian motion. Architectonics, a term borrowed from the philosophical literature, symbolizes the eventual union of the structure of theories-how our minds construct the world-with the theory of structures-or how stability is maintained in the chaotic world of microsystems.
|
Click on image below to
access full-text publication.
 |
Proteins 2000; 41:374-384.
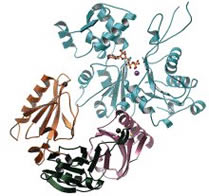
A Comparative Structural Analysis of the ADF/Cofilin Family.
Bowman GD, Nodelman IM, Chua NH, Lindberg U, Schutt CE
Actin-depolymerizing factor (ADF) and cofilin define a family of actin-binding proteins essential for the rapid turnover of filamentous actin in vivo. Here we present the 2.0 Å crystal structure of Arabidopsis thaliana ADF1 (AtADF1), the first plant crystal structure from the ADF/cofilin (AC) family. Superposition of the four AC isoform structures permits an accurate sequence alignment that differs from previously reported data for the location of vertebrate-specific inserts and reveals a contiguous, vertebrate-specific surface opposite the putative actin-binding surface. Extending the structure-based sequence alignment to include 30 additional isoforms indicates three major groups: vertebrates, plants, and other eukaryotes. Within these groups, several structurally conserved residues that are not conserved throughout the entire AC family have been identified. Residues that are highly conserved among all isoforms tend to cluster around the tryptophan at position 90 and a structurally conserved kink in a-helix 3. Analysis of surface character shows the presence of a hydrophobic patch and a highly conserved acidic cluster, both of which include several residues previously implicated in actin binding. |
Click on image below to
access full-text publication.
 |
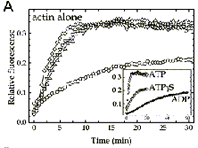
FEBS Lett. 2000; 476 (3):155-159.
Covalent Binding of ATPgS to the Nucleotide-Binding Site in S14C-actin.
Schuler H, Schutt CE, Lindberg U, Karlsson R
We have recently reported on the characterization of beta-actin carrying the mutation S14C in one of the phosphate-binding loops. The present paper describes the attachment of the adenosine 5'-[g-thio]-triphosphate (ATPgS) to actin containing this mutation. Treatment of S14C-actin with ATPgS blocked further nucleotide exchange and raised the thermal stability of the protein, suggesting the formation of a covalent bond between the sulfhydryl on the terminal phosphate of ATPgammaS and cysteine-14 of the mutant actin. The affinity of the derivatized G-actin for DNase I as compared to wild-type ATP-actin was lowered to a similar extent as that of ADP.AlF(4)-actin. The derivatized actin polymerized slower than ATP-actin but faster than ADP-actin. Under these conditions the bound ATPgammaS was hydrolyzed, suggesting the formation of a state corresponding to the transient ADP.P(i)-state. ATPgS-actin interacted normally with profilin, whereas the interaction with actin depolymerizing factor (ADF) was disturbed, as judged on the effects of these proteins on actin polymerization. |
Click on image below to
access full-text publication.
 |
Eur. J. Biochem. 2000; 267 (13): 4054-4062.
Mutational Analysis of Arginine 177 in the Nucleotide Binding Site of β-Actin.
Schuler H, Nyakern M, Schutt CE, Lindberg U, Karlsson R
Actin ADP-ribosylated at arginine 177 is unable to hydrolyze ATP, and the R177 side chain is in a position similar to that of the catalytically essential lysine 71 in heat shock cognate protein Hsc70, another member of the actin-fold family of proteins. Therefore, actin residue R177 has been implicated in the mechanism of ATP hydrolysis. This paper compares wild-type beta-actin with a mutant in which R177 has been replaced by aspartic acid. The mutant beta-actin was expressed in Saccharomyces cerevisiae and purified by DNase I-affinity chromatography. The mutant protein exhibited a reduced thermal stability and an increased nucleotide exchange rate, suggesting a weakened interdomain connection. The ATPase activity of G-actin and the ATPase activity expressed during polymerization were unaffected by the R177D replacement, showing that this residue is not involved in catalysis. In the presence of polymerizing salts, ATP hydrolysis by both wild-type Mg-beta-actin and the mutant protein preceded filament formation. With the mutant actin, the initial rate of ATP hydrolysis was as high as with wild-type actin, but polymer formation was slower, reached lower steady-state levels, and the polymers formed exhibited much lower viscosity. The critical concentration of polymerization (Acc) of the mutant actin was increased 10-fold as compared to wild-type actin. Filaments formed from the R177D mutant beta-actin bound phalloidin. |
Click on image below to
access full-text publication.
 |
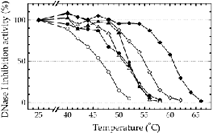
Eur. J. Biochem. 2000; 267 (2): 476-486.
Thermal Unfolding of G-actin Monitored with the DNase I-inhibition Assay; Stabilities of Actin Isoforms.
Schuler H, Lindberg U, Schutt CE, Karlsson R
Actin is one of the proteins that rely on chaperonins for proper folding. This paper shows that the thermal unfolding of G-actin, as studied by CD and ultraviolet difference spectrometry, coincides with a loss in DNase I-inhibiting activity of the protein. Thus, the DNase I inhibition assay should be useful for systematic studies of actin unfolding and refolding. Using this assay, we have investigated how the thermal stability of actin is affected by either Ca2 + or Mg2 + at the high affinity divalent cation binding site, by the concentration of excess nucleotide, and by the nucleotide in different states of phosphorylation (ATP, ADP.Pi, ADP. Vi, ADP.AlF4, ADP.BeFx, and ADP). Actin isoforms from different species were also compared, and the effect of profilin on the thermal stability of actin was studied. We conclude that the thermal unfolding of G-actin is a three-state process, in which an equilibrium exists between native actin with bound nucleotide and an intermediate free of nucleotide. Actins in the Mg-form were less stable than the Ca-forms, and the stability of the different isoforms decreased in the following order: rabbit skeletal muscle alpha-actin = bovine cytoplasmic gamma-actin > yeast actin > cytoplasmic beta-actin. The activation energies for the thermal unfolding reactions were in the range 200-290 kJ.mol- 1, depending on the bound ligands. Generally, the stability of the actin depended on the degree with which the nucleotide contributed to the connectivity between the two domains of the protein. |
Click on image below to
access full-text publication.
 |
J. Mol. Biol. 1999; 294 (5): 1271-1285.
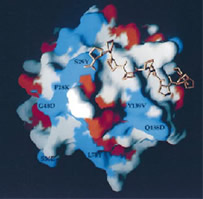
X-ray Structure Determination of Human Profilin II: A Comparative Structural Analysis of Human Profilins.
Nodelman IM, Bowman GD, Lindberg U, Schutt CE
Human profilins are multifunctional, single-domain proteins which directly link the actin microfilament system to a variety of signalling pathways via two spatially distinct binding sites. Profilin binds to monomeric actin in a 1:1 complex, catalyzes the exchange of the actin-bound nucleotide and regulates actin filament barbed end assembly. Like SH3 domains, profilin has a surface-exposed aromatic patch which binds to proline-rich peptides. Various multidomain proteins including members of the Ena/VASP and formin families localize profilin:actin complexes through profilin:poly-L-proline interactions to particular cytoskeletal locations (e.g. focal adhesions, cleavage furrows). Humans express a basic (I) and an acidic (II) isoform of profilin which exhibit different affinities for peptides and proteins rich in proline residues. Here, we report the crystallization and X-ray structure determination of human profilin II to 2.2 Å. This structure reveals an aromatic extension of the previously defined poly-L-proline binding site for profilin I. In contrast to serine 29 of profilin I, tyrosine 29 in profilin II is capable of forming an additional stacking interaction and a hydrogen bond with poly-L-proline which may account for the increased affinity of the second isoform for proline-rich peptides. Differential isoform specificity for proline-rich proteins may be attributed to the differences in charged and hydrophobic residues in and proximal to the poly-L-proline binding site. The actin-binding face remains nearly identical with the exception of five amino acid differences. These observations are important for the understanding of the functional and structural differences between these two classes of profilin isoforms. |
Click on image below to
access full-text publication.
 |
Les Cahiers de Science & Vie 1999; 53: 78-87.
Nouveau Regard Sur le Muscle.
Kreatsoulas C, Lindberg U, Schutt CE
C'est par le mouvement coordonné de milliers de molécules que les muscles produisent des forces. La simulation, sur ordinateur, de ce "ballet" moléculaire amène à formuler une nouvelle théorie de leur contraction. |
Click on image below to
access full-text publication.

|
Eur. J. Biochem. 1999; 265 (1): 210-220.
Mutational Analysis of Ser14 and Asp157 in the Nucleotide-Binding Site of β-actin.
Schuler H, Korenbaum E, Schutt CE, Lindberg U, Karlsson R
This paper compares wild-type and two mutant beta-actins, one in which Ser14 was replaced by a cysteine, and a second in which both Ser14 and Asp157 were exchanged (Ser14-->Cys and Ser14-->Cys, Asp157-->Ala, respectively). Both of these residues are part of invariant sequences in the loops, which bind the ATP phosphates, in the interdomain cleft of actin. The increased nucleotide exchange rate, and the decreased thermal stability and affinity for DNase I seen with the mutant actins indicated that the mutations disturbed the interdomain coupling. Despite this, the two mutant actins retained their ATPase activity. In fact, the mutated actins expressed a significant ATPase activity even in the presence of Ca2+ ions, conditions under which actin normally has a very low ATPase activity. In the presence of Mg2+ ions, the ATPase activity of actin was decreased slightly by the mutations. The mutant actins polymerized as the wild-type protein in the presence of Mg2+ ions, but slower than the wild-type in a K+/Ca2+ milieu. Profilin affected the lag phases and elongation rates during polymerization of the mutant and wild-type actins to the same extent, whereas at steady-state, the concentration of unpolymerized mutant actin appeared to be elevated. Decoration of mutant actin filaments with myosin subfragment 1 appeared to be normal, as did their movement in the low-load motility assay system. Our results show that Ser14 and Asp157 are key residues for interdomain communication, and that hydroxyl and carboxyl groups in positions 14 and 157, respectively, are not necessary for ATP hydrolysis in actin. |
Click on image below to
access full-text publication.
 |
Acta Physiol. Scand. 1998;163 (4): 307-323.
Muscle Contraction as a Markov Process. I: Energetics of the Process.
Schutt CE, Lindberg U
Force generation during muscle contraction can be understood in terms of cyclical length changes in segments of actin thin filaments moving through the three-dimensional lattice of myosin thick filaments. Recent anomalies discovered in connection with analysis of myosin step sizes in in vitro motility assays and with skinned fibres can be rationalized by assuming that ATP hydrolysis on actin accompanies these length changes. The paradoxically rapid regeneration of tension in quick release experiments, as well as classical energetic relationships, such as Hill's force-velocity curve, the Fenn effect, and the unexplained enthalpy of shortening, can be given mutually self-consistent explanations with this model. When muscle is viewed as a Markov process, the vectorial process of chemomechanical transduction can be understood in terms of lattice dependent transitions, wherein the phosphate release steps of the myosin and actin ATPases depend only on occurrence of allosteric changes in neighbouring molecules. Tropomyosin has a central role in coordinating the steady progression of these cooperative transitions along actin filaments and in gearing up the system in response to higher imposed loads.
|
Click on image below to
access full-text publication.
 |
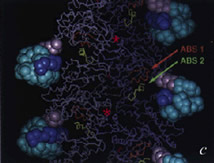
J. Mol. Biol. 1998; 280 (3): 463-474.
Domain Motions in Actin.
Page R, Lindberg U, Schutt CE
Previous crystallographic investigations have shown that actin can undergo large conformational changes, even when complexed to the same actin binding protein. We have conducted a formal analysis of domain motions in actin, using the four available crystal structures, to classify the mechanism as either hinge or shear and to quantify the magnitude of these changes. We demonstrate that actin consists of two rigid cores, a semi-rigid domain and three conformationally variable extended loops. Confirming predictions about the nature of the domain rotation in actin based on its structural similarity to hexokinase, we show, using an algorithm previously used only to identify protein hinges, that residues at the interface between the two rigid cores undergo a shear between alternative conformations of actin. Rotations of less than 7 degrees in the torsion angles of five residues in the polypeptides that connect the rigid cores enable one actin conformation to be transformed into another. Because these torsion angle changes are small, the interface between the domains is maintained. In addition, we show that actin secondary structure elements, including those outside the rigid cores, are conformationally invariant among the four crystal structures, even when actin is complexed to different actin binding proteins. Finally, we demonstrate that the current F-actin models are inconsistent with the principles of actin conformational change identified here. |
Click on image below to
access full-text publication.
 |
Nat. Struct. Biol. 1997; 4 (3): 169-72.
Plugging into Actin's Architectonic Socket.
Schutt CE, Kreatsoulas C, Page R, Lindberg U
"Plugging into the 'architectonic socket' might initiate a flow of momentum along actin filaments and through the extended structures that contribute to cortical tension."
|
Click on image below to
access full-text publication.
 |
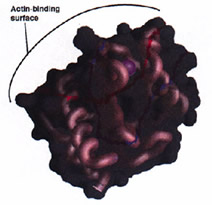
Structure 1997; 5 (1): 19-32.
The Crystal Structure of a Major Allergen from Plants.
Thorn KS, Christensen HE, Shigeta R, Huddler D, Shalaby L, Lindberg U, Chua NH, Schutt CE
The determination of the Arabidopsis thaliana profilin isoform I structure, using multiwavelength anomalous diffraction (MAD) to obtain structure-factor phases, is reported here. The structure of Arabidopsis profilin is similar to that of previously determined profilin structures. Conserved amino acid residues in profilins from plants, mammals, and lower eukaryotes are critically important in dictating the geometry of the PLP-binding site and the overall polypeptide fold. The main feature distinguishing plant profilins from other profilins is a solvent-filled pocket located in the most variable region of the fold. CONCLUSIONS: Comparison of the structures of SH3 domains with those of profilins from three distinct sources suggests that the mode of PLP binding may be similar. A comparison of three profilin structures from different families reveals only partial conservation of the actin-binding surface. The proximity of the semi-conserved actin-binding site and the binding pocket characteristic of plant profilins suggests that epitopes encompassing both features are responsible for the cross-reactivity of antibodies between human and plant profilins thought to be responsible for type I allergies. |
Click on image below to
access full-text publication.
 |
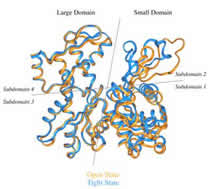
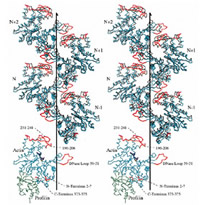
J. Mol. Biol. 1996; 263 (4): 607-623.
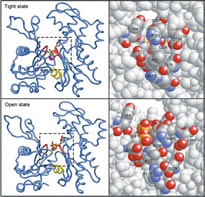
The Structure of an Open State of β-actin at 2.65 Å Resolution.
Chik JK, Lindberg U, Schutt CE
The structure of an "open state" of crystalline profilin:beta-actin has been solved to 2.65 Å by X-ray crystallography. The open-state crystals, in 1.8 M potassium phosphate, have an expanded unit cell dimension in the c direction of 185.7 Å compared with 171.9 Å in the previously solved ammonium sulphate-stabilized "tight-state" structure. The unit cell change between the open and the tight states is accompanied by large subdomain movements in actin. Furthermore, the nucleotide in the open state is significantly more exposed to solvent, and local conformational changes in the hydrophobic pocket surrounding cysteine 374 occur during the transition to the tight state. Significant changes were observed at the N terminus and in the DNase-I binding loop. Neither the structure of profilin nor its contact with beta-actin are affected by the changes in the unit cell. Applying osmotic pressure to profilin:beta-actin crystals brings about a collapse of the unit cell comparable with that seen in the open to tight-state transition, enabling an estimate of the work required to cause this transformation of beta-actin in the crystals. The slight difference in energy between the open and collapsed states explains the extreme sensitivity of profilin:beta-actin crystals to changes in chemical and thermal environment. |
Click on image below to
access full-text publication.
 |
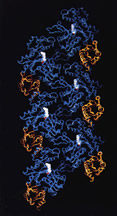
| J. Struct. Biol. 1995; 115 (2): 186-198.
A Discourse on Modeling F-Actin.
Schutt CE, Rozycki MD, Myslik JC, Lindberg U
A major challenge for structural biologists is to obtain atomic resolution models for higher order structures. In favorable instances, the intact particle can be crystallized and all atomic positions and bonds within the macromolecular complex can be determined to the resolution limit of the data. Icosahedrally symmetric virus particles exemplify the application of crystallography to structures of polymeric complexes at sufficient resolution to visualize the amino-acid side chain interactions stabilizing intermolecular surfaces (Harrison et al., 1978). Tobacco mosaic virus is an outstanding example of the successful phasing of a high-resolution fiber X-ray diffraction pattern by multiple isomorphous replacement (Namba & Stubbs, 1986). But, what can be when crystals of the intact structures cannot be obtained or when the existing fiber patterns simply do not extend to high enough resolution? |
Click on image below to
access full-text publication.
|
| Biophys. J. 1995; 68(4 Suppl): 12S-17S; discussion 17S-18S.
Structural Studies on the Ribbon-to-Helix Transition in Profilin: Actin Crystals.
Schutt CE, Rozycki MD, Chik JK, Lindberg U
Knowledge of the structure of actin in its various conformational states is important for understanding the diverse motile activities carried out by eukaryotic cells. Profilin:actin crystals provide a unique system for studying conformational states of actin, because they exhibit a high degree of polymorphism in response to environmental conditions while maintaining crystalline order. A preliminary comparison of two states of profilin:beta-actin crystals shows that crystal polymorphism involves movements of actin subdomains at hinge points homologous to those found in hexokinase, a protein whose polypeptide fold is related to actin. The homology of the hinge points in actin to those in hexokinase suggests that actin subdomain movements in profilin:beta-actin crystals have functional significance. We discuss how these movements could be related to structural transitions between states of filamentous actin in muscle contraction. |
Click on image below to
access full-text publication.

|
Nature 1993; 365: 810-816.
The Structure of Crystalline Profilin- β-actin.
Schutt CE, Myslik JC, Rozycki MD, Goonesekere NC, Lindberg U
The three-dimensional structure of bovine profilin- β-actin has been solved to 2.55 Å resolution by X-ray crystallography. There are several significant local changes in the structure of β-actin compared with alpha-actin as well as an overall 5 degrees rotation between its two major domains. Actin molecules in the crystal are organized into ribbons through intermolecular contacts like those found in oligomeric protein assemblies. Profilin forms two extensive contacts with the actin ribbon, one of which appears to correspond to the solution contact in vitro.
|
Click on image below to
access full-text publication.
 |
FEBS Lett. 1993; 325 (1-2): 59-62.
A New Perspective on Muscle Contraction.
Schutt CE, Lindberg U
Recent experimental findings suggest that the myosin cross-bridge theory may no longer be adequate to account for certain basic facts concerning muscle contraction. A newly-proposed mechanism based on length changes in actin filaments might be the basis for a simpler explanation for how the free energy of ATP hydrolysis can be transduced into work by muscle fibers. |
Proc. Nat. Acad. Sci. U.S.A. 1992; 89 (1): 319-323.
Actin as the Generator of Tension During Muscle Contraction.
Schutt CE, Lindberg U
We propose that the key structural feature in the conversion of chemical free energy into mechanical work by actomyosin is a myosin-induced change in the length of the actin filament. As reported earlier, there is evidence that helical actin filaments can untwist into ribbons having an increased intersubunit repeat. Regular patterns of actomyosin interactions arise when ribbons are aligned with myosin thick filaments, because the repeat distance of the myosin lattice (429 Å) is an integral multiple of the subunit repeat in the ribbon (35.7 Å). This commensurability property of the actomyosin lattice leads to a simple mechanism for controlling the sequence of events in chemical-mechanical transduction. A role for tropomyosin in transmitting the forces developed by actomyosin is proposed. In this paper, we describe how these transduction principles provide the basis for a theory of muscle contraction. |
Click on image below to
access full-text publication.
 |
|