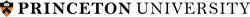

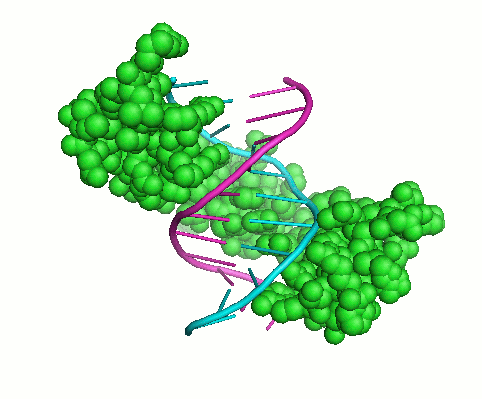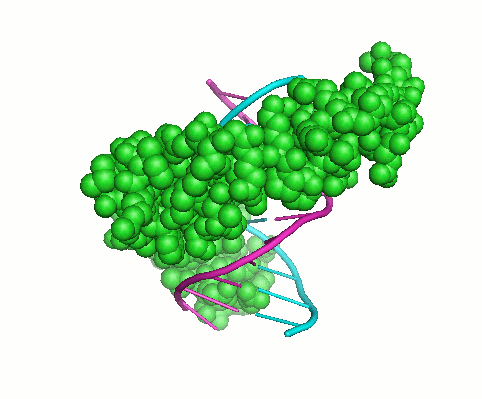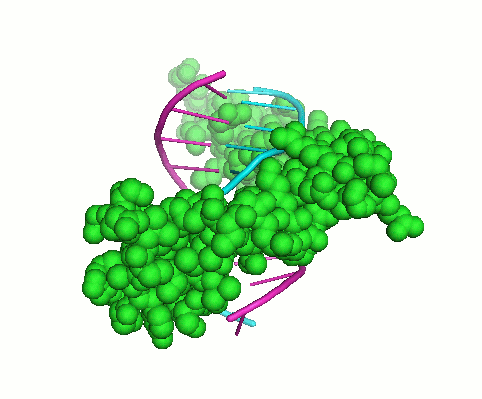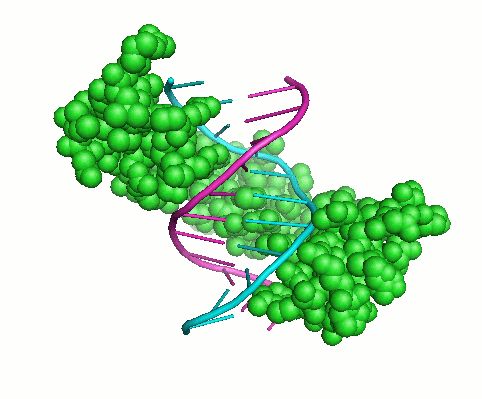
Collagen Triple-Helix
The increasing family of collagens and proteins with collagenous domains shows the collagen triple-helix, consisting of three supercoiled polyproline-II helices, to be one of the major protein motifs. The distinguishing feature of the collagen-like sequence is the presence of Gly as every third residue, resulting in Gly-X-Y repeating pattern, where X and Y denote a generic amino acid and may be occupied by any residue, most frequently by prolines. During my work in Barbara Brodsky’s lab, we have evaluated basic principles of collagen stability. This includes computational and experimental determination of individual amino acid propensities for X and Y-positions (1) and evaluation of amino acid interactions in triple-helical context (2,3). This work resulted in the development, for the first time, an algorithm for accurate prediction of collagen stability directly from amino acid sequence. Recently updated interactive tool, Collagen Stability Calculator (4), is available for prediction whether an input sequence will form a collagen triple-helix and calculates its thermal stability. Detailed studies of triple-helical 3D structures led us to designing short synthetic collagen-like peptides capable to self-associate and form fibrils.Specific Aims: Understanding of genotype – phenotype relationships
Mutations in human collagen genes, resulting in breaking the repeating Gly-X-Y pattern in collagen sequence, lead to a number of human disorders, including brittle bone disease, aortic aneurism and skin abnormalities. However, the molecular basis of why single mutation may leads to various phenotypes is understood poorly. It was previously proposed that not all Gly mutations in collagens result in pathology (5). The availability of a substantial dataset of mutations in collagens makes possible to relate them to local properties of the collagen triple-helix, including its stability and functionality. Understanding of molecular mechanisms for mutations in collagens will lead to a rational treatment of these serious disorders. My current goal here is to understand why the same mutation, e.g. Gly->Ser, may or may not result in pathology, depending on the local amino acid sequence surrounding the mutation site. This will form the basis for NIH grant proposal. The hypothesis is to be tested by molecular modeling, machine learning and peptide approaches.
1. Molecular Simulations. The stability variations in collagens will be calculated using Collagen Stability Calculator to determine a distribution of stable domains along collagen molecule. The molecular dynamics simulations will be applied to estimate the effect of clinically observed mutations on equilibrium and dynamic propertied of corresponding collagen domains. The effect of pathological mutations will be evaluated in terms of structural disturbances, including variations of dihedral angles and break-up of interchain hydrogen bonding.
2. Synthetic Peptide Models. It is hypothesized here that the level of collagen destabilization determines the clinical outcome of collagen mutations. Molecular dynamics simulations should help to pre-select the most interesting collagen mutations from clinically observed database. Corresponding normal and mutated collagen regions will be incorporated in new improved synthetic peptide models. The effect of mutations on folding and stability will be thermodynamically evaluated by the combination of biophysical techniques.
3. Developing 3D collagen model. The recent availability of full-length collagen structure provides an opportunity to evaluate spatial models for collagen interaction and binding. Collagens are known to bind many ligands and self-associate in a specific manner. Unidimensional collagen interaction map was previously designed by Dr. James San Antonio to visualize all known functional sites along with clinically observed mutations along the collagen sequence. I am currently collaborating with Dr. San Antonio on designing spatial collagen map. The new map will allow more accurate 3D positioning of all the functional / mutation sites and mapping out the accessible surface of the collagen fibrils with D-period. It will be estimated, which regions of the fibril are exposed and thus relevant to cell and ligand interactions. This is expected to lead to better understanding of collagen self-association mechanisms, interactions with ligands and the genotype-phenotype relationships in collagen disorders.
4. Prediction of pathological phenotypes. Machine learning is an artificial intelligence method to automatically produce models from data, when practical model formulation is impossible to human brain. It is proposed here to apply the Principal Component Analysis and the Support Vector Machine to learn molecular factors determining pathological phenotypes at collagen mutations. A number of different factors were proposed to have such an effect, including local stability, domain organization, location of functional site, amino acid variations etc. These molecular factors when listed together with phenotypic outcome will be used as a training set. Machine learning will lead to (1) determination of the molecular factors which have the most significant effect on phenotype and (2) an accurate prediction of phenotypic outcome from the knowledge of amino acid mutations in collagens.
References: