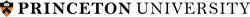

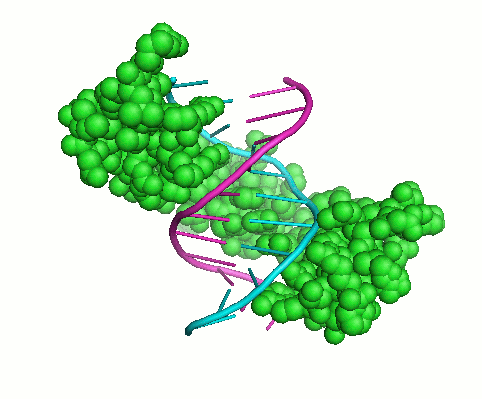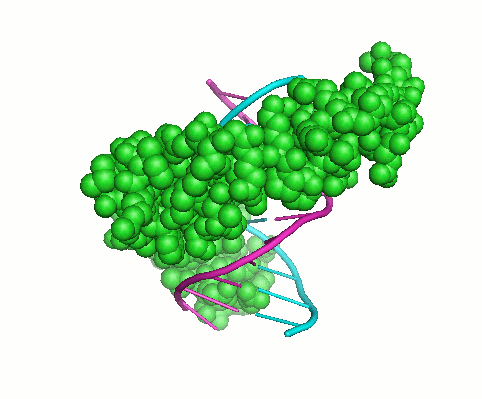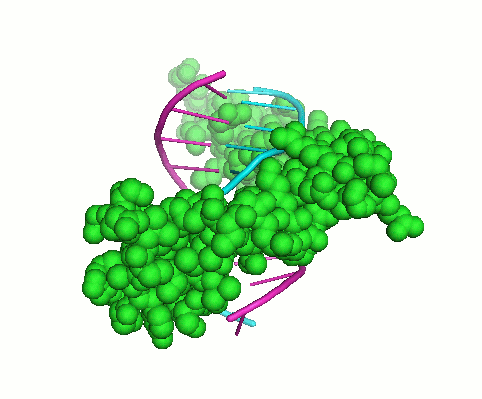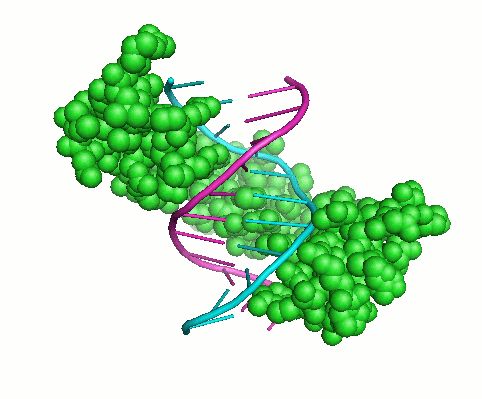
Cys2His2 Zinc Finger Domain
Regulatory proteins are frequently having modular structure, consisting from domains that mediate molecular interaction and other activities. This makes them somewhat similar to collagens when relatively short well-conserved protein motif determines the specificity of interactions of large molecules. These small domains are often good targets for drugs. Computational approaches can be developed for predicting the binding preferences of such protein motifs, even when no binding sites are known for a particular protein. The Cys2His2 zinc finger proteins represent the largest class of eukaryotic transcription factors. The DNA binding interface of each of zinc fingers (ZF) consists of only four amino acid-nucleotide contacts per zinc finger domain. We recently built a high-quality literature-derived experimental database of ZF–DNA binding examples (Persikov et all. 2009). On the basis of this database, we trained a support vector machine (SVM), a machine learning technique, to predict ZF protein – DNA binding, which outperforms previous computational approaches. The great advantage of this approach is that SVM can naturally incorporate constraints about the relative binding affinities of protein-DNA pairs (the information not previously used). This project resulted in developing an interactive tool for predicting ZF protein- DNA binding.Specific Aims: Structural Interface for ZF protein – DNA binding
Prominently, the training of SVM can also lead to evaluation of protein – DNA binding interfaces. Optimum structural model for Cys2His2 protein – DNA binding is highly debated in the last decade. We found that SVM based on polynomial kernels outperforms linear SVM when trained on the basis of four-contact canonical model. This indicates the presence of dependencies between contacts in the canonical binding model and suggests that modification of the underlying structural model may result in further improved performance in predicting ZF protein-DNA binding. Experimental information about non-binding and relative binding of protein-DNA pairs will be used to evaluate all possible interfaces for Cys2His2 protein – DNA binding. Accurate structural binding model will lead to better understanding of interaction mechanism and result in more effective prediction of protein–DNA interactions. There is a sufficient background and enough experimental data to propose another NIH grant on this topic. Structure analysis and machine learning methods will be applied to uncover structural information from conventional binding experiments.
1. Co-crystal structure analysis. There are over 20 high-resolution Cys2His2 ZF protein – DNA crystal structures in Protein Data Bank. These structures will be analyzed to determine which amino acid – base pairs have a potential to form interacting pairs.
2. Machine learning approach. All possible structural models will be evaluated by SVM. The analysis of pre-trained SVM classifier (weight vector) allows ranking the importance of amino acid – base contacts listed in structural binding model. Using the previously collected high-confidence experimental database, different SVMs will be trained on the basis of different structural binding models (from simplest canonical model to all-possible-contact model). The best performing SVM model will point out an optimum structural binding model for Cys2His2 protein – DNA binding.
3. Experimental evaluation. Protein and DNA sequences predicted to form a complex with a novel binding interface will be purified/synthesized and tested to form a complex using various binding assays. The dissociation constants will be measured for the novel complexes and compared with canonical binding complexes (Zif268 protein). The isothermal titration calorimetry will be used for thermodynamic evaluation of the Cys2His2 protein – DNA binding.