
Hush, little virus, don’t say a word: How Princeton scientists investigate sleeping viruses
Princeton scientists are investigating how viruses become latent in the nervous system -- and how to prevent it. Here, cell bodies (gold, with blue-stained nuclei) and axons (green) show the effects of being infected with a virus (red) for 24 hours.
Worldwide, four in five adults are infected with herpes simplex virus (HSV), say researchers. But most of those infected don’t show symptoms like cold sores because the virus infection is “latent” or “silent” — sleeping — within the nervous system, where it remains for the entire life of its host.
While many virus researchers are interested in understanding what causes these sleeping viruses to wake up, or reactivate, Princeton scientists are trying to understand what prevents the infection from going to sleep in the first place.
To uncover how herpes virus infections become silent and what triggers escape from silencing, several researchers have attempted to create simplified in vitro model systems using neuronal cells that are cultured in petri dishes and infected with viruses.
Unfortunately, in vitro virus infections are nearly always in their productive phase — awake — so to induce latency, researchers have used harsh methods, explained Orkide Ozge Koyuncu, an associate research scholar in molecular biology. The typical approach requires dissociating the target cells and dosing them with interferon or a replication inhibitor prior to virus infection.
As sleep studies go, it’s like clubbing your subjects and then hoping that the bodies on the floor will wake up normally.
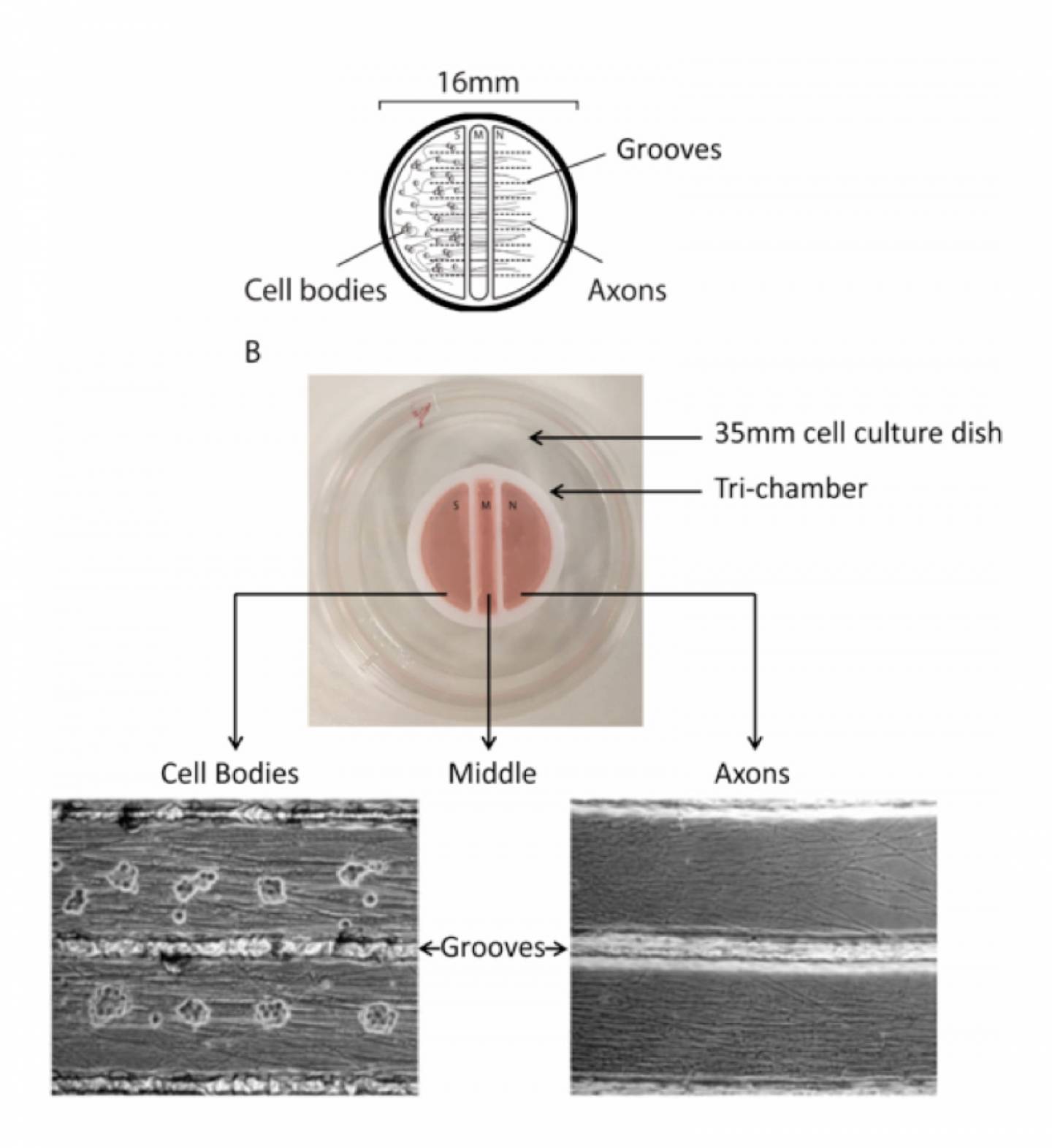
The modified Campenot tri-chamber is 16 millimeters across and consists of a cell body (S) compartment, a middle (M) compartment and a neurite (N) compartment. Parallel grooves are etched on the plastic surface to guide growing axons to the M and N compartments. In the center image, an actual tri-chamber is set on a 35-mm cell culture dish. At the bottom are microscopic views showing tightly packed rat SCG neurons in the S compartment and pure axons bundled together between the grooves of the N compartment.
But Koyuncu found a gentler approach — more like singing them a lullaby — that allowed her to lull viruses to sleep in cultured neurons without the use of any inhibitors.
Together with her colleagues Margaret MacGibeny, an M.D./Ph.D. student, then-postdoctoral researcher Ian Hogue, and Lynn Enquist, the lab’s principal investigator and the Henry L. Hillman Professor in Molecular Biology, Koyuncu has used this unique latency model system to reveal specific cellular and viral components that can prevent the infection from going to sleep. Their results appear in the Oct. 26 issue of PLoS Pathogens.
This new approach “represents an important advancement,” said Felicia Goodrum Sterling, the director of the graduate program in immunology at the University of Arizona Cancer Center, who was not involved in the research.
“In understanding herpesvirus latency, model systems are everything,” Goodrum Sterling explained. “An important, overarching goal of the field is to develop culture systems using primary cells with minimal ex vivo [external] manipulation. ... This is the first model system that does not require drug treatment to establish latency.”
Koyuncu and her colleagues use a three-compartment system to culture neurons that preserves the basic structure — the “architecture” — of the nerves that these viruses target. In humans, individual nerves can be surprisingly long, with the longest running from your toes to your spine. At one end, the nerves reach the skin cells where they gather sensory information, and at the other end is the cell body, which contains the nucleus and the neuron’s genetic information. These neuronal cell bodies are protected deep within the body. In between is the axon, the highway-like path along which information is carried back and forth between the peripheral tissues and the nervous system.
Part of what makes herpes viruses so interesting is their ability to traverse this axonal highway, said Koyuncu. Infection happens in peripheral tissues, then the virus particles enter axons and zoom into the distant cell nucleus, where the viral genetic information can become latent, sleeping for days or even years. When the genomes “wake up,” new virus particles are made and these can travel along the axon back out to the peripheral tissue, which creates the cold sores that can spread infection to new hosts. In rare cases, virus particles can travel to the brain and cause encephalitis.
When neurons are dissociated, which has been the standard approach for studying latency until now, the cell bodies with their axons are jumbled together, making it impossible to mimic the natural route of nervous system invasion via isolated axons.
In Koyuncu’s approach, a divided cell chamber called a modified Campenot tri-chamber allows researchers to keep the axons and cell bodies separated. “This is a simplified system,” she said. “I like it because you not only can control the viral dose infecting axons but also are able to monitor each step — from transport in axons to expression in the nucleus — by live-cell microscopy.”
Once the neurons, seeded on one side of the chamber, grew axons into the other two chambers, Koyuncu introduced a small number of virus particles to isolated axons. This step is sufficient to naturally lull the infection to sleep in the nuclei of the cell bodies without the need for interferon or viral replication inhibitors. The cell bodies recognize that they are infected and activate a defense system to silence the incoming viral genetic information.
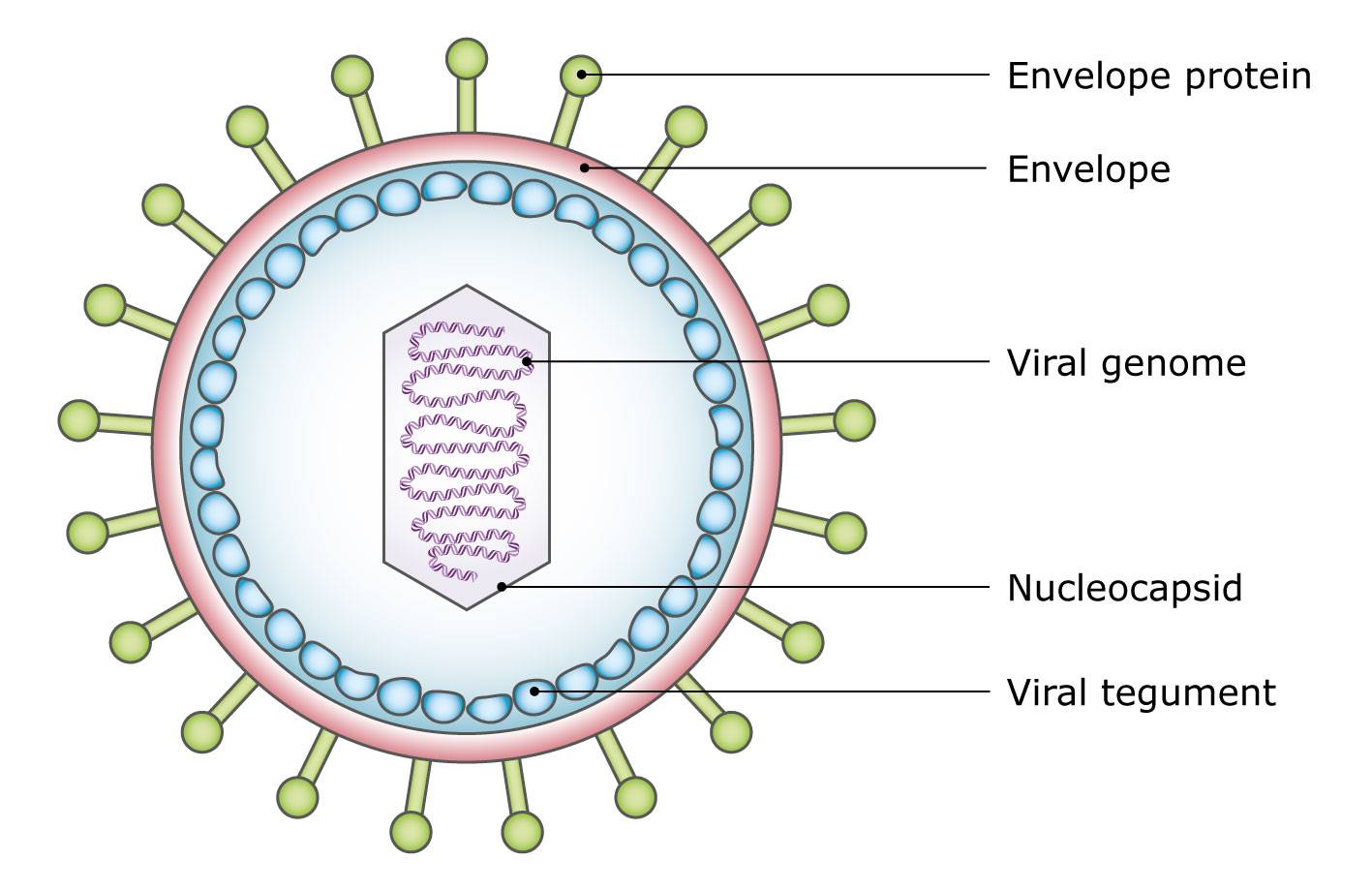
A viral tegument, also known as a viral matrix, is the cluster of proteins lining the envelope that surrounds the herpes virus core. Koyuncu and her colleagues identified two ways for herpes viruses to escape from silencing: a stress-mediated slow route initiated by the neuron, and a fast route activated by these viral tegument proteins.
Once the infection could be dependably lulled to sleep, Koyuncu tackled a new question: Can viruses “escape” from this initial silencing fate? What processes or pathways enable virus genomes to remain in a productive infection mode where genomes are expressed, and viral proteins and new virus particles are produced?
When herpes viruses invade the nervous system, viral DNA is recognized as foreign. Neuronal cells shut off viral gene expression to protect the nervous system, but the invading virus genetic material remains, silently waiting in neurons for a trigger to wake up and produce more active virus.
So far, Koyuncu and her colleagues have identified two ways for herpes viruses that are destined to be silenced to escape from silencing: a stress-mediated slow route that is initiated by the neuron, and a fast route that is activated by viral tegument proteins. A viral tegument, also known as a viral matrix, is the cluster of proteins lining the envelope that surrounds the herpes virus core.
The slow process is initiated by the neurons when the host experiences stress, like sunburn, hormonal changes or physical trauma. The neurons begin producing proteins to protect themselves. Once killed, a neuron cannot be replaced.
The stress signals wake up the silent virus infection, which begins producing a few new viral proteins — but not enough to be considered a productive infection, explained Enquist.
“These viral proteins are necessary, but not sufficient, to reliably start a productive infection,” he said. Something more was needed: the fast process that Koyuncu and her colleagues discovered.
The fast process depends on a set of viral proteins that the researchers believe “supercharges” the activation of the silent viral information. Koyuncu discovered this when she infected isolated axons with a small number of virus particles, which would normally travel to the cell bodies and be silenced, while simultaneously exposing the cell bodies to a large number of inactivated virus particles that have been dosed with ultraviolet radiation (UV).
This had the effect of pouring a bucket of ice water on sleepy viruses to keep them awake once they reach the nucleus.
Although ultraviolet radiation renders the viral genome unable to reproduce, the researchers found that these irradiated particles, which still contain tegument proteins and broken genomes, were sufficient to keep the virus awake.
This was her first big “aha!” moment, almost two years ago, Koyuncu recalled: “By infecting the cell bodies with virus particles that were inactivated by UV and simultaneously infecting axons with viruses that would normally be silenced, I saw a productive infection in three days — which was really fast. I said, ‘OK, now we have a robust system to study sleep/wake signals!’”
“This was amazing,” said Enquist, “but the question was then, ‘What are the viral components that promote escape from silencing?’”
One popular hypothesis, explained MacGibeny, was that a large number of viral genomes could saturate the silencing machinery in the nucleus, essentially “swamping out the neuron’s defense system.”
But she and Koyuncu instead infected cell bodies with what they called “light particles” — virus particles that had been hollowed out, leaving the envelope and tegument proteins but with no genome or core — and found that they were able to achieve the same escape from silencing.
“These experiments with the ‘light particles’ — with no genomes — suggest that excess genomes are not required for escape from silencing, whereas viral tegument proteins are critical,” she said.
The Princeton team also found that when viral tegument proteins are present in the cell body, escape from silencing does not require previously identified stress signals.
As a medical student, MacGibeny looks at the research through a less theoretical lens than her colleagues. “If you can understand the molecular mechanisms underlying how latency is established and how viral genomes can escape from silencing, then you can start to identify some particular therapeutic targets that we could eventually go after [with drugs].”
Enquist agreed and gave the bottom line: “We identified the class of viral proteins that supercharge the neuronal stress response to wake up sleeping herpes virus genomes.”
The work was supported by the National Institutes of Health (grants RO1 NS33506 and NS060699 to LWE) and the National Institute of Neurological Disorders and Stroke (F30 NS090640 Fellowship to MM).
