Artificial intelligence detects a new class of mutations behind autism
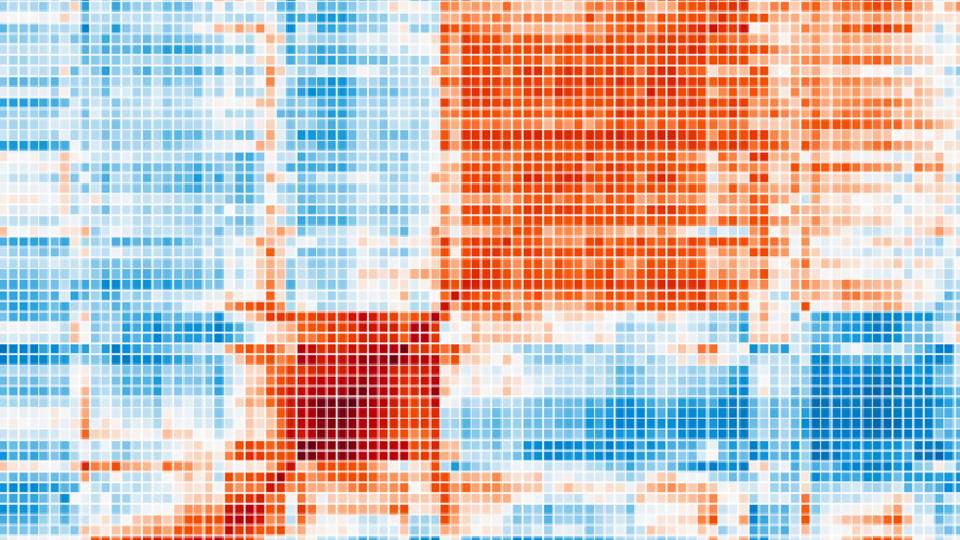
Research led by Olga Troyanskaya, a Princeton professor of computer science and genomics, has led to a new method for searching the entire human genome for mutations that affect how genes are regulated and predicting how those mutations might be associated with diseases such as autism. Genes predicted to be disrupted by regulatory mutations in people with autism tended to be involved in brain cell functioning and fell into two categories. One category (shown in blue) relates to synapses, communication hubs between neurons, and the other (shown in red) relates to chromatin, the highly structured form of DNA and proteins required for proper gene expression in chromosomes.
Many mutations in DNA that contribute to disease are not in actual genes but instead lie in the 99% of the genome once considered "junk." Even though scientists have recently come to understand that these vast stretches of DNA do in fact play critical roles, deciphering these effects on a wide scale has been impossible until now.
Using artificial intelligence, a Princeton University-led team has decoded the functional impact of such mutations in people with autism. The researchers believe this powerful method is generally applicable to discovering such genetic contributions to any disease.
Publishing May 27 in the journal Nature Genetics, the researchers analyzed the genomes of 1,790 families in which one child has autism spectrum disorder but other members do not. The method sorted among 120,000 mutations to find those that affect the behavior of genes in people with autism. Although the results do not reveal exact causes of cases of autism, they reveal thousands of possible contributors for researchers to study.
Much previous research has focused on identifying mutations in genes themselves. Genes are essentially instructions for making the many proteins that build and control the body. Mutations in genes result in mutated proteins whose functions are disrupted. Other types of mutations, however, disrupt how genes are regulated. Mutations in these areas affect not what genes make but when and how much they make.
Until now, it was not possible to look across the entire genome for snippets of DNA that regulate genes and to predict how mutations in this regulatory DNA are likely to contribute to complex disease, the researchers said. This study is the first proof that mutations in regulatory DNA can cause a complex disease.
"This method provides a framework for doing this analysis with any disease," said Olga Troyanskaya, professor of computer science and genomics and a senior author of the study. The approach could be particularly helpful for neurological disorders, cancer, heart disease and many other conditions that have eluded efforts to identify genetic causes.
"This transforms the way we need to think about the possible causes of those diseases," said Troyanskaya, who also is deputy director for genomics at the Simons Foundation's Flatiron Institute in New York where she led a group of co-authors.
The team also included a group led by neuroscientist Robert Darnell of The Rockefeller University. The first authors of the paper are Jian Zhou and Christopher Park, who earned Ph.D.s at Princeton and are now visiting collaborators at the Lewis-Sigler Institute for Integrative Genomics and researchers at the Flatiron Institute, and Chandra Theesfeld at Princeton’s Lewis-Sigler Institute for Integrative Genomics.
Most previous research on the genetic basis of disease has focused on the 20,000 known genes and the surrounding sections of DNA that regulate those genes. However, even this enormous amount of genetic information makes up only slightly more than 1% of the 3.2 billion chemical pairs in the human genome. The other 99% has conventionally been thought of as "dark" or "junk," although recent research has begun to disrupt that idea.
In their new finding, the research team offers a method to make sense of this vast array of genomic data. The system uses an artificial intelligence technique called deep learning in which an algorithm performs successive layers of analysis to learn about patterns that would otherwise be impossible to discern. In this case, the algorithm teaches itself how to identify biologically relevant sections of DNA and predicts whether those snippets play a role in any of more than 2,000 protein interactions that are known to affect the regulation of genes. The system also predicts whether disrupting a single pair of DNA units would have a substantial effect on those protein interactions.
The algorithm "slides along the genome" analyzing every single chemical pair in the context of the 1,000 chemical pairs around it, until it has scanned all mutations, Troyanskaya said. The system can thus predict the effect of mutating each and every chemical unit in the entire genome. In the end, it reveals a prioritized list of DNA sequences that are likely to regulate genes and mutations that are likely to interfere with that regulation.
Prior to this computational achievement, the conventional way to glean such information would be painstaking laboratory experiments on each sequence and each possible mutation in that sequence. This number of possible functions and mutations is too big to contemplate — an experimental approach would require testing each mutation against more than 2,000 types of protein interactions and repeating those experiments over and over across tissues and cell types, amounting to hundreds of millions of experiments. Other research groups have sought to accelerate this discovery by applying machine learning to targeted sections of DNA, but had not achieved the ability to look at each DNA unit and each possible mutation and the effects on each of more than 2,000 regulatory interactions across the whole genome.
"What our paper really allows you to do is take all those possibilities and rank them," said Park. "That prioritization itself is very useful, because now you can also go ahead and do the experiments in just the highest priority cases."
Lastly, the system calibrates its predictions based on known disease-causing mutations and develops a "disease impact score," an assessment of how likely a given mutation is to have an effect on disease.
In the case of autism, the researchers analyzed the genomes of 1,790 families with "simplex" autism spectrum disorder, meaning the condition is apparent in one child but not in other members of the family. (These data were taken from the Simons Simplex Collection of more than 2,000 autism families.) Among this sample, fewer than 30% of the people affected by autism spectrum disorder had a previously identified genetic cause. The newly found mutations are likely to significantly increase that fraction, the researchers said.
The ability to predict the functional effect of each mutation was the key innovation in this new study. Previous studies had found it challenging to detect any difference in the number of regulatory mutations in people with autism compared to unaffected people. The new method, however, looked at mutations predicted to have a high functional impact, and found a significantly higher number of such mutations in affected people.
When the researchers then looked at what genes were affected by these mutations, they turned out to be genes strongly associated with brain functions. These newly discovered mutations affected similar genes and functions as do previously identified mutations.
"Now we open the field to understand all the factors that may be involved in autism," said Theesfeld.
This information also is important to families and their doctors to better diagnose the disorder and to avoid making overly general assumptions how one person's autism might be classified with others. "They say that when you meet one person with autism you have met one person with autism because no cases are alike," said Theesfeld. "Genetically, it seems to be the same way."
With this new method, the team is analyzing the genetic causes of various forms of cancer, heart disease and other disorders.
The work was funded by the NIH and Simons Foundation. Other co-authors of the study include Aaron Wong, Julien Funk and Kevin Yao of Flatiron, and Yuan Yuan, Claudia Scheckel, John Fak and Yoko Tajima of Rockefeller.